Агонист-антагонист — Agonist-antagonist — qaz.wiki
Агонист против антагонистаВ фармакологии термин агонист-антагонист или смешанный агонист / антагонист используется для обозначения лекарственного средства, которое в одних условиях ведет себя как агонист (вещество, которое полностью активирует рецептор, с которым оно связывается), а в других условиях ведет себя как антагонист ( вещество, которое связывается с рецептором, но не активируется и может блокировать активность других агонистов).
Типы смешанных агонистов / антагонистов включают лиганды рецепторов, которые действуют как агонисты для некоторых типов рецепторов и антагонисты для других, или агонисты в одних тканях, в то время как антагонисты в других (также известные как селективные модуляторы рецепторов ).
Синаптические рецепторы
Для синаптических рецепторов агонист — это соединение, которое увеличивает активацию рецептора, связываясь непосредственно с ним или увеличивая количество времени, в течение которого нейротрансмиттеры находятся в синаптической щели.
На альфа — адренорецепторов , ( R ) -3-nitrobiphenyline представляет собой α 2C селективный агонист, а также является слабым антагонистом на & alpha ; 2А и & alpha ; 2В подтипов.
Опиоиды-агонисты-антагонисты
Наиболее известными агонистами-антагонистами являются опиоиды . Примеры таких опиоидов:
Опиоиды-агонисты-антагонисты обычно имеют максимальный эффект — при приеме определенной дозы они не увеличивают свою эффективность. Следовательно, опиоиды-агонисты-антагонисты имеют более низкий потенциал привыкания, но также более низкую анальгетическую эффективность и с большей вероятностью вызывают психотомиметические эффекты.
Опиоиды-агонисты-антагонисты, которые блокируют дельта при активации мю-опиоидных рецепторов, вызывают анальгезию без развития толерантности .
Смотрите также
Ссылки
<img src=»https://en.wikipedia.org//en.wikipedia.org/wiki/Special:CentralAutoLogin/start?type=1×1″ alt=»» title=»»>Агонист
Пользователи также искали:

…
(PDF) Инверсированные агонисты: больше, чем антагонисты?
10
мор и неосознанный страх, т.е. сим-
птомы тревожного состояния. При
комбинированном применении нейро-
активных соединений инверсирован-
ный агонист этих рецепторов RO 19-
4603 был эффективным антагонистом
этанола, устраняя характерные ло-
комоторные нарушения и снижая по-
требление этанола у линии алкоголь-
предпочитающих крыс [11].
Опиоидные рецепторы. Хрони-
ческое введение морфина увеличива-
ет конститутивную активность мю-
опиоидных рецепторов, что позво-
ляет исследовать эффекты инверси-
рованных агонистов. Налоксон, обла-
дающий такими свойствами, снижа-
ет спонтанную активацию опиоидных
рецепторов, что приводит к возник-
новению абстинентного синдрома [12].
Дофаминовые и серотониновые
рецепторы. Галоперидол проявля-
ет свойства полного инверсирован-
ного агониста дофаминовых рецепто-
ров, в то время как клозапин – лишь
частичного. Однако в отношении се-
ротониновых 5НТ2С рецепторов ати-
пичные антипсихотические средства
(клозапин, оланзапин, рисперидон)
являются инверсированными агони-
стами, а типичные нейролептики –
нейтральными антагонистами. Таким
образом, различия между типичны-
ми и атипичными антипсихотически-
ми средствами состоят не только в
спектре, но и характере взаимодей-
ствия с рецепторами. Многообещаю-
щие данные получены в отношении
других подтипов серотониновых ре-
цепторов. Так, инверсированный аго-
нист 5-HT1В рецепторов SB-224289
облегчал обучение эксперименталь-
ных животных, при этом антагони-
сты 5-НТ1В рецепторов ослабляли его
действие [13].
Ангиотензиновые рецепторы.
Конститутивная активация харак-
терна для АТ1 рецепторов. Механиче-
ские растяжения мышцы сердца при-
водят к спонтанной активации этих
рецепторов без участия ангиотензи-
на. Лазартан, кандесартан и олме-
сартан проявляют свойства инверси-
рованных агонистов и способны сни-
зить экспрессию генов, а также реак-
цию протеинкиназы на внешние сти-
мулы [14].
Адренорецепторы. В исследова-
ниях на изолированных миоцитах,
полученных у пациентов с сердеч-
ной недостаточностью, было показа-
но, что карведилол и метопрополол
обладают свойствами инверсирован-
ных агонистов в отношении бета1-
рецепторов, в то время как буцин-
долол проявляет свойства их анта-
гониста. При клинических испыта-
ниях карведилол и метопролол сни-
жали смертность пациентов от сер-
дечной недостаточности, а буциндо-
лол нет. Одним из возможных объ-
Одним из возможных объ-
яснений может быть наличие мута-
ции S49G бета1-рецепторов у паци-
ентов с сердечной недостаточностью.
При этой мутации частота спонтан-
ной активации рецепторов возраста-
ет. Способность угнетать спонтанную
рецепторную активность инверсиро-
ванными агонистами приводит к по-
ложительному клиническому эффек-
ту [15].
Еще одним возможным клиниче-
ским применением инверсированных
агонистов бета-адренорецепторов мо-
жет стать лечение бронхиальной аст-
мы. Современные бета-2 адреноми-
метики, используемые как бронхо-
литики, со временем вызывают эф-
фект down-регуляции рецепторов,
что приводит к снижению эффек-
тивности лечения. Инверсирован-
ные агонисты (надолол) при хрониче-
ском применении увеличивают число
постсинаптических рецепторов, что
может быть использовано при лече-
нии бронхиальной астмы [16].
Виды протоколов ЭКО | Полезное от клиники «Геном» в Ростове на Дону
Протокол ЭКО — это схема введения препаратов для стимуляции работы яичников в режиме сверх нормы, чтобы получить качественные яйцеклетки для оплодотворения. Это так называемая стимуляция суперовуляции.
Основными считаются ДВА вида протоколов ЭКО — короткий и длинный. Также, используются протоколы ЭКО с антагонистами.
Первый этап ЭКО определяет судьбу протокола. Если, в результате стимуляции суперовуляции, будут получены качественные яйцеклетки, то шанс на получение беременности в итоге программы ЭКО возрастает. Квалификация и опыт репродуктологов «Геном» позволяют разрабатывать индивидуальные схемы стимуляции суперовуляции, которые позволяют получить адекватный ответ яичников у женщин как «до», так и «после» 35 лет, а также у пациенток с низким фолликулярным запасом, при наличии таких заболеваниях как эндометриоз, миома матки, кисты яичников.
Основа любой схемы стимуляции суперовуляции – гонадотропины. Это препараты, которые содержат гормоны ФСГ (фолликулостимулирующий) или ЛГ (лютеинизирующий).
Это препараты, которые содержат гормоны ФСГ (фолликулостимулирующий) или ЛГ (лютеинизирующий).
Некоторые содержат только один ФСГ — например, «Гонал» и «Пурегон», а некоторые — комбинации ЛГ и ФСГ, например, «Менопур». Для предупреждения преждевременной овуляции используют агонисты или антагонисты гонадотропин-рилизинг гормона (ГнРГ).Агонисты ГнРГ обладают схожим действием с ГнРГ. При ежедневном введении они раздражают гипофиз, вызывая чрезмерную выработку гормонов ЛГ и ФСГ. В результате, гипофиз прекращает вырабатывать собственные гормоны, наступает состояние, похожее на климакс. Это даёт врачу возможность полностью «руководить» дальнейшим процессом стимуляции. Агонисты ГнРГ применяются иногда в качестве триггера овуляции – это снижает риск такого осложнения, как гиперстимуляция яичников. Антагонисты ГнРГ действуют противоположно ГнРГ. Они подавляют секрецию гормонов гипофиза сильнее агонистов, но, при этом, активность гипофиза после прекращения их приема восстанавливается быстрее.
 Дополнительные преимущества антагонистов перед агонистами:
Дополнительные преимущества антагонистов перед агонистами: — возможно сокращение длительности стимуляции на 1-2 дня,
— рост фолликулов происходит быстрее,
— требуется меньшая доза препаратов (снижается гормональная нагрузка),
— качество эмбрионов и, следовательно, шанс на имплантацию и наступление беременности выше.
1) «Короткий» с агонистами ГнРГ
Короткий протокол оправдывает своё название тем, что проводится в одном менструальном цикле. Здесь меньше медикаментозная нагрузка на организм женщины и риск возникновения СГЯ, но есть вероятность спонтанной овуляции. Яйцеклетки созревают неравномерно, поэтому, их может быть недостаточно или они будут ненадлежащего качества.
2) «Длинный» с агонистами ГнРГ
Длинный протокол захватывает 2 цикла и может длиться до 50 дней. Начинается он с регулирующей фазы (обычно на 21 день цикла), в результате чего происходит полная остановка выработки собственных гормонов. Далее, вводятся искусственные гормоны по схеме, назначенной репродуктологом. При этом, процесс полностью «подвластен» специалисту, риск самопроизвольной овуляции сведён к нулю. В итоге, как правило, получают достаточное количество зрелых яйцеклеток.
Далее, вводятся искусственные гормоны по схеме, назначенной репродуктологом. При этом, процесс полностью «подвластен» специалисту, риск самопроизвольной овуляции сведён к нулю. В итоге, как правило, получают достаточное количество зрелых яйцеклеток.
Гиперстимуляция яичников является результатом роста большого количества фолликулов. Их стенки активно продуцируют эстрогены, избыточный уровень этих гормонов в крови приводит к соответствующим реакциям — сгущению крови, появлению жидкости в полостях (грудной, брюшной). Лёгкая форма СГЯ, которая неизбежна в некоторых ситуациях, не нарушает общего состояния. Пациентка может ощущать дискомфорт в области живота, лёгкое недомогание. Эти симптомы достаточно быстро корректируется без каких либо серьёзных последствий для здоровья.3) Протоколы с антагонистами могут применяться для женщин разного возраста, при различных формах бесплодия. Репродуктологи отмечают, что частота наступления беременности в них выше, при этом гормональную нагрузку можно свести к минимуму. Поэтому, такие программы ЭКО применяются всё чаще.

★ Частичный агонист — фармакология .. Информация
★ Частичный агонист
В фармакологии термин частичный агонист используется в отношении лекарств и химических соединений, которые являются лигандами для специфических подтипов клеточных рецепторов и способен активировать рецептор, то есть перевести его в активный пространственной конфигурации, но с меньшей вероятностью, чем эндогенный агонист того же рецептора, рецепторов, эффективность которых принято за 100 % и который рассматривается, следовательно, как полный агонист. иными словами, агонист активности частичного агониста, по определению, всегда больше 0 % но меньше, чем 100 %.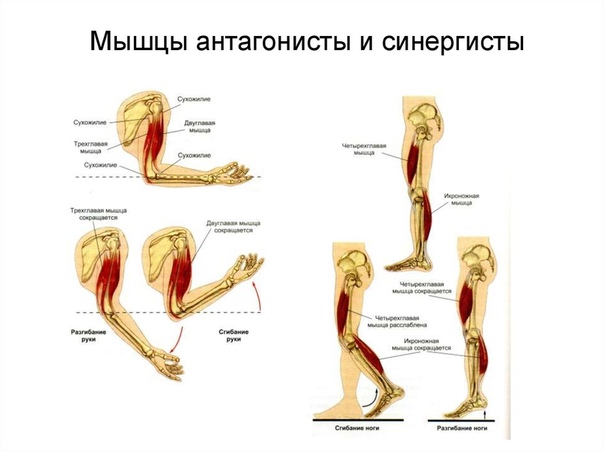
На практике, внутренней агонистической активности соединения, как правило, рассматривается как «частичных агонистов» как правило, выше 10-20 %, но ниже 70-80 % как «слабые» частичного агониста с внутренней агонистической активностью, меньшей 10-20 % и в эксперименте и в клинической практике обычно трудно отличить от «истинных» нейтральные антагонисты строго ноль внутренней агонистической активностью, «сильные» частичного агониста с внутренней агонистической активностью, большей 70-90 % трудно отличить от «истинных» полные агонисты имея внутренней агонистической активностью, строго равна 100 %. кроме того, там, на самом деле, очень мало «истинных» нейтральные антагонисты со строго нулевой внутренней агонистической активностью — большинство из них либо слабые и очень слабые частичные агонисты или обратных агонистов. как раз таки там очень мало «истинных» полные агонисты в дополнение к эндогенным агонистом, которая по определению принимается за 100 % — большинство из них просто очень сильный или сильный частичные агонисты. кроме того, даже если эксперимент на некоторые соединения, полученное значение внутренней агонистической активностью, строго равна 0 % или 100 %, это не значит, что эта смесь действительно «истинным нейтральным антагонистом» или «истинным полным агонистом» — это просто означает, что разница между измеренным значением и 0 % или 100 % — это меньше, чем погрешность метода измерения. таким образом, с формальной математической точки зрения, частичные агонисты являются наиболее распространенным типом экзогенных лигандов, и в зависимости от величины внутренней агонистической активностью клинически они могут быть рассмотрены и применены в качестве квази- «антагонистов» слабые частичные агонисты с внутренней агонистической активностью по крайней мере 10-20 % на активность эндогенных лигандов или как квази- «полных агонистов» сильная частичного агониста с внутренней агонистической активностью выше 70-90 % на активность эндогенных лигандов или как «частичных агонистов» при промежуточных значениях внутренней агонистической активностью.
кроме того, даже если эксперимент на некоторые соединения, полученное значение внутренней агонистической активностью, строго равна 0 % или 100 %, это не значит, что эта смесь действительно «истинным нейтральным антагонистом» или «истинным полным агонистом» — это просто означает, что разница между измеренным значением и 0 % или 100 % — это меньше, чем погрешность метода измерения. таким образом, с формальной математической точки зрения, частичные агонисты являются наиболее распространенным типом экзогенных лигандов, и в зависимости от величины внутренней агонистической активностью клинически они могут быть рассмотрены и применены в качестве квази- «антагонистов» слабые частичные агонисты с внутренней агонистической активностью по крайней мере 10-20 % на активность эндогенных лигандов или как квази- «полных агонистов» сильная частичного агониста с внутренней агонистической активностью выше 70-90 % на активность эндогенных лигандов или как «частичных агонистов» при промежуточных значениях внутренней агонистической активностью.
Частичные агонисты могут также рассматриваться в качестве лигандов, которые показывают, как агонистические и антагонистические свойства в зависимости от конкретной клинической или экспериментальной ситуации, или, другими словами, как «смешанные агонисты-антагонисты». А именно, когда биологические системы, в то же время частичные агонисты и полный агонист, например, эндогенным агонистом или частичным агонистом сильнее тех же рецепторов, «более слабый» частичный агонист, на самом деле, проявляет свойства конкурентным антагонистом этих рецепторов, соревнуясь с «более сильным» частичного агониста или полный агонист, в том числе эндогенным лигандом для трудоустройства рецепторы и вызывая общее снижение уровня активности рецепторного системы по сравнению с наличием одного лишь только полного агониста или «более сильного» частичный агонист в концентрации. клинического применения и эффективность частичных агонистов определяются тем, что они могут одновременно активации рецепторного аппарата в низком уровне стимуляции низкий уровень эндогенного полный агонист к желаемому «субмаксимального» уровень которых ниже, чем при использовании полного агониста, и, чтобы предотвратить чрезмерную, вредные и гиперстимуляция рецепторов, происходит на слишком высоком уровне эндогенного агониста. способность частичных агонистов действовать как конкурентные антагонисты в присутствии полного агониста в том числе эндогенным лигандом или в присутствии «более сильного» частичный агонист является крайне важным для клинической практики. так, например, способность налоксона для снятия симптомов опиоидной интоксикации на основе этого свойства. не менее важное значение для клинической практики способность сильной частичного агониста с рецептором эффективности 80-90 % и выше, чтобы действовать почти ничем не отличается от «истинных» полные агонисты. так, например, Прессорное вещество фенилэфрин мезатон, который является структурным аналогом норадреналина, на самом деле довольно сильная, «почти полным» частичные агонисты α-адренорецепторов, а не «истинным» полный агонист. но разница настолько мал, что не имеет клинического значения и позволяет использовать фенилэфрин как «почти полного агониста», Прессорных веществ для купирования гипотонии короткодействующие и неудобное в использовании норадреналина.
способность частичных агонистов действовать как конкурентные антагонисты в присутствии полного агониста в том числе эндогенным лигандом или в присутствии «более сильного» частичный агонист является крайне важным для клинической практики. так, например, способность налоксона для снятия симптомов опиоидной интоксикации на основе этого свойства. не менее важное значение для клинической практики способность сильной частичного агониста с рецептором эффективности 80-90 % и выше, чтобы действовать почти ничем не отличается от «истинных» полные агонисты. так, например, Прессорное вещество фенилэфрин мезатон, который является структурным аналогом норадреналина, на самом деле довольно сильная, «почти полным» частичные агонисты α-адренорецепторов, а не «истинным» полный агонист. но разница настолько мал, что не имеет клинического значения и позволяет использовать фенилэфрин как «почти полного агониста», Прессорных веществ для купирования гипотонии короткодействующие и неудобное в использовании норадреналина. аналогично, сальбутамол сильно, «почти полным» частичные агонисты β-адренергических рецепторов настолько сильна, что клинически его действие на бронхи не отличается от действия адреналина, который позволяет использовать его в качестве бронхолитического средства.
аналогично, сальбутамол сильно, «почти полным» частичные агонисты β-адренергических рецепторов настолько сильна, что клинически его действие на бронхи не отличается от действия адреналина, который позволяет использовать его в качестве бронхолитического средства.
Другие важные примеры препаратов, которые являются частичные агонисты определенных рецепторов и в истинном «сбалансированном» в смысле — не в смысле схожести выше экстремальные примеры с налоксоном и fenilafrinom и сальбутамол включают nebenzodiazepinova анксиолитик буспирон, нетиповой антипсихотик арипипразол, частичные агонисты опиоидных рецепторов наркотический анальгетик бупренорфин, метаболит клозапина norclozapine. есть также примеры лигандов, которые активируют рецептор PPAR (НПСР) γ как частичный агонист — хонокиол и falcarinol.
Арипипразол – механизм действия и место в стратегии применения антипсихотиков нового поко ления
Арипипразол – новый атипичный антипсихотик с механизмом действия, который качественно отличает его от представленных на рынке атипичных антипсихотических препаратов.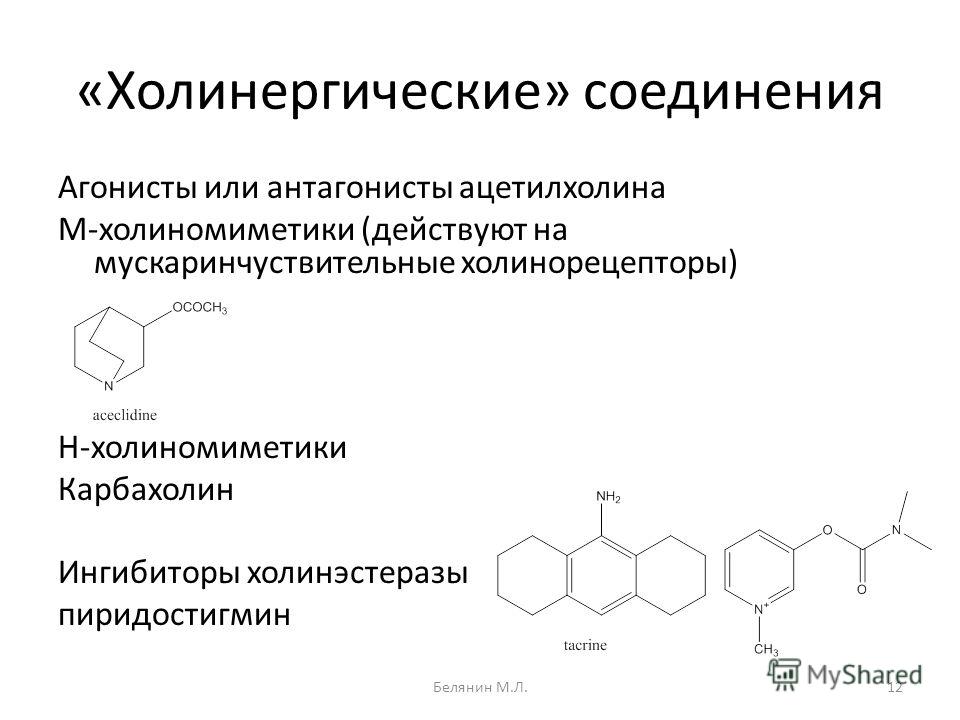 Он демонстрирует одновременно свойства агониста и антагониста на моделях дофаминергической активности и гиперактивности соответственно. Препарат относится к числу первых антипсихотиков, механизм действия которых непосредственно направлен как на негативную, так и на позитивную симптоматику шизофрении; его экстрапирамидные побочные эффекты слабо выражены, а риск набора веса и седации минимален; он не вызывает удлинения интервала QT и повышения уровня пролактина.
Он демонстрирует одновременно свойства агониста и антагониста на моделях дофаминергической активности и гиперактивности соответственно. Препарат относится к числу первых антипсихотиков, механизм действия которых непосредственно направлен как на негативную, так и на позитивную симптоматику шизофрении; его экстрапирамидные побочные эффекты слабо выражены, а риск набора веса и седации минимален; он не вызывает удлинения интервала QT и повышения уровня пролактина.
Частичный агонизм к дофамину и функциональная селективность являются инновационными стратегиями в фармакологическом лечении шизофрении и аффективных расстройств. Эти стратегии позволили совершить прорыв в понимании модуляции дофаминергических структур, которая при прежнем подходе заключалась преимущественно в антагонизме к рецепторам дофамина типа D2(D2R). Несмотря на тот факт, что арипипразол был включен в терапевтические схемы более 12 лет назад, многие вопросы, которые касаются сложности воздействия этого средства на передачу сигнала и внутриклеточные процессы, обусловленные его плейотропным рецепторным профилем, до сих пор остаются открытыми.
Описание механизма действия этого препарата из-за его комплексности постепенно сместилось от частичного агонизма к функциональной селективности. Было доказано, что арипипразол влияет на множественные межклеточные процессы и связи, от индукции ранних генов до модуляции поддерживающих белков и активации факторов транскрипции, а также на некоторые кортикальные и подкорковые процессы нейротрансмиссии. Все больше данных подтверждают, что, кроме оккупации D2-рецепторов, арипипразол обладает уникальным нейробиологическим действием, отличающим его от всех доступных антипсихотических препаратов. Особенно подчеркивается его влияние при долгосрочном применении на сродство к D2-рецепторам и их количество; проводится соответствующий анализ воздействия препарата при длительной терапии психоза. Предполагаемые эффекты арипипразола на клеточно-защитные механизмы и рост нейритов, а также его дифференциальное воздействие на внутриклеточные процессы (с участием киназы, регулируемой внеклеточными сигналами [extracellular signal regulated kinase, ERK]) по сравнению с эффектами полных антагонистов D2-рецепторов требуют дальнейшего исследования. Целью настоящего обзора является описание основных нейробиологических эффектов арипипразола и анализ потенциальных возможностей терапии шизофрении благодаря более широкому спектру дофаминергической модуляции, а не исключительно антагонизму D2-рецепторов.
Целью настоящего обзора является описание основных нейробиологических эффектов арипипразола и анализ потенциальных возможностей терапии шизофрении благодаря более широкому спектру дофаминергической модуляции, а не исключительно антагонизму D2-рецепторов.
Прошло уже более 40 лет с момента выдвижения гипотезы о роли дофаминовой дисфункции в патофизиологии шизофрении. Данная теория остается центральной, несмотря на то, что некоторые доклинические и клинические исследования доказывают влияние и других нейротрансмиттеров, прежде всего глутаматергических и серотонинергических. Дофаминергическая гипотеза утверждает, что увеличение высвобождения дофамина в стриатуме связано со снижением дофаминергического тонуса в дорсальной префронтальной коре. Подобная механистическая интерпретация кажется слишком упрощенной, особенно с учетом сложности молекулярной нейробиологии шизофрении, которая в последнее время рассматривается как нарушение синаптической пластичности и высвобождения дофамина.
Ключевые аспекты:
- Атипичный антипсихотик арипипразол обладает уникальным фармакологическим профилем, который обеспечивает «адаптивную» фармакологическую активность.
- В зависимости от уровня эндогенного дофамина и активности передачи в дофаминергических путях арипипразол может действовать как полный антагонист, умеренный антагонист или частичный агонист D2-рецепторов дофамина (D2R).
- Эффективность арипипразола в основном объясняется комбинацией частичного агонизма/антагонизма в отношении D2R и 5-HT1A серотониновых рецепторов вместе с антагонизмом в отношении 5-HT2A серотониновых рецепторов.
-
Однако рецепторный профиль арипипразола намного сложнее, а исследования на животных моделях показали, что препарат влияет на несколько типов межклеточной передачи, некоторые корковые и подкорковые нейротрансмиттерные системы, а также оказывает воздействие на экспрессию генов, в отличие от других антипсихотиков.

- С учетом фармакологических и функциональных характеристик арипипразола становятся возможными новые механизмы воздействия на дофаминергические структуры для лечения психоза, что позволяет улучшить качество терапии шизофрении и аффективных расстройств.

Многочисленные данные, начиная с доклинических моделей и заканчивая нейровизуализационными исследованиями у людей in vivo, демонстрируют нарушенное высвобождение дофамина в стриатуме пациентов с шизофренией. В связи с этим динамические исследования по измерению связывания 11C-раклоприда со стриарными дофаминовыми D2-рецепторами (D2R) и дофаминовыми D3-рецепторами (D3R) у пациентов с шизофренией и здоровых контрольных испытуемых после острой провокации амфетамином убедительно доказали, что шизофрения ассоциируется с более значимым высвобождением дофамина в стриатуме и что подобное чрезмерное высвобождение может коррелировать с позитивными симптомами.
В контексте дофаминовой гипотезы сложность функции рецептора дофамина заслуживает особого внимания, поскольку до появления арипипразола все доступные антипсихотики характеризовались блокадой D2R (хотя и в разной степени), а соединения, лишенные этой способности, не обладали эффективным антипсихотическим действием. Появление арипипразола изменило этот взгляд и позволило впервые задействовать в лечении психоза клинически значимый механизм, основанный на оккупации D2R без их блокады. Доклинические исследования и исследования с участием пациентов in vivo, помимо специфического действия арипипразола на D2R, продемонстрировали его отличительные эффекты на дофаминергическую передачу сигналов и ряд эффекторов транскрипции (по сравнению с действием антипсихотиков – полных антагонистов D2R, блокирующих данные рецепторы). Эти эффекты варьируют от селективной индукции ранних генов до модуляции различных поддерживающих белков (белков скэффолда) и факторов транскрипции, а также позволяют предположить, что действие вещества направлено на множественные межклеточные связи и процессы, что обусловливает терапевтическое действие на ряд нейробиологических функций, не свойственное обычным антипсихотикам нового поколения. Благодаря такому механизму действия арипипразол можно комбинировать с другими нейролептиками. Это обеспечивает дополнительный клинический эффект, в том числе при вторичной резистентности к антипсихотической терапии, развившейся в процессе лечения.
Благодаря такому механизму действия арипипразол можно комбинировать с другими нейролептиками. Это обеспечивает дополнительный клинический эффект, в том числе при вторичной резистентности к антипсихотической терапии, развившейся в процессе лечения.
Фармакокинетические свойства арипипразола
вверх
Абсорбция и распределение
После приема внутрь средний период полувыведения арипипразола составляет около 75 часов, а активного метаболита дегидроарипипразола – 94 часа. Время достижения устойчивой концентрации – 14 дней после начала применения. Концентрация в плазме достигает пика в течение 3-5 часов после приема таблетки. Биодоступность препарата в форме таблеток составляет 87%, а плазменные концентрации выше таковых при использовании раствора для перорального приема. Прием с пищей, особенно с высоким содержанием жиров, не влияет на среднюю пиковую концентрацию в плазме (Cmax) или площадь под кривой «концентрация в плазме – время», при этом удлиняя среднее время сохранения максимальной концентрации арипипразола в сыворотке (Tmax) примерно на 3 часа. Равновесный объем распределения высокий, что указывает на обширное внесосудистое распределение. В терапевтических концентрациях как арипипразол, так и дегидроарипипразол на 99% связываются с сывороточными белками, главным образом альбумином. Возраст, пол и масса тела не влияют на фармакокинетику препарата. Полная занятость D2R наблюдалась при плазменных концентрациях арипипразола, превышающих 100-150 нг/мл у больных и 100-200 нг/мл у здоровых лиц. Купирование симптомов было более существенным у пациентов с сывороточными концентрациями от 150 до 300 нг/мл.
Равновесный объем распределения высокий, что указывает на обширное внесосудистое распределение. В терапевтических концентрациях как арипипразол, так и дегидроарипипразол на 99% связываются с сывороточными белками, главным образом альбумином. Возраст, пол и масса тела не влияют на фармакокинетику препарата. Полная занятость D2R наблюдалась при плазменных концентрациях арипипразола, превышающих 100-150 нг/мл у больных и 100-200 нг/мл у здоровых лиц. Купирование симптомов было более существенным у пациентов с сывороточными концентрациями от 150 до 300 нг/мл.
Метаболизм и выведение
Арипипразол метаболизируется в основном изоферментами CYP3A4 и CYP2D6 цитохрома P450 (CYP) человека до дегидроарипипразола и некоторых других метаболитов. В свою очередь, дегидроарипипразол метаболизируется только CYP3A4 и CYP2D6 до нескольких компонентов, полученные метаболиты выводятся с мочой или калом. Из-за таких метаболических особенностей применение арипипразола вместе с ингибиторами CYP3A4 или CYP2D6 может потенциально влиять на фармакокинетику соединения.
Кроме того, было установлено, что системный клиренс арипипразола сильно снижается под действием селективных ингибиторов обратного захвата серотонина (СИОЗС) пароксетина (мощного ингибитора CYP2D6) и флувоксамина (менее мощного ингибитора как CYP2D6, так и CYP3A4). При этом пароксетин демонстрировал более высокий результат, чем флувоксамин, на что значительное влияние оказывал генотип испытуемых. Эти наблюдения имеют большое значение, поскольку арипипразол часто используется в комбинации с антидепрессантами в лечении психотической депрессии или депрессивных симптомов при шизофрении. Арипипразол также часто применяется одновременно со стабилизаторами настроения для лечения биполярного расстройства. В одном исследовании было отмечено, что совместное применение арипипразола и карбамазепина (индуктора активности CYP3A4) снизило значения Cmax и площади под кривой «концентрация в плазме – время» для арипипразола и дегидроарипипразола.
Структура лиганд-рецепторного взаимодействия, присущая механизму действия арипипразола
вверх
В настоящее время основное определение функций агонистов/антагонистов и препаратов/лигандов расширено за счет включения таких понятий, как обратный агонизм и смещенная передача сигнала. Эти и другие механизмы взаимодействия препарат-мишень имеют фундаментальное значение в области разработки лекарственных средств.
Эти и другие механизмы взаимодействия препарат-мишень имеют фундаментальное значение в области разработки лекарственных средств.
Краткое описание свойств связывания препарата приводится для введения основных понятий, которые будут способствовать описанию механизма действия арипипразола.
Фармакологические эффекты, обусловленные взаимодействием препарата с его рецепторными мишенями, в основном определяются уникальными свойствами препарата:
- сродство к мишени;
- эффективность или внутренняя активность;
- селективность;
- механизм взаимодействия;
- время удержания.
Сродство препарата характеризует способность молекулы связываться с ее биологической мишенью (т. е. рецептором, ферментом, транспортной системой). Для агониста (или антагониста) численное представление сродства, обозначаемое KA (или KB для антагонистов), выражено обратным значением равновесной константы диссоциации (KD) комплекса лиганд-рецептор.
Внутренняя активность определяет способность лиганда инициировать реакцию на молекулярном, клеточном, тканевом или системном уровне и зависит от свойства молекулы занимать и активировать (агонист) или блокировать (антагонист) свою мишень. Идея «внутренней активности» лиганд-рецепторного взаимодействия обусловила такую классификацию лигандов: полные агонисты, частичные агонисты, нейтральные антагонисты и обратные агонисты. Полным агонизмом называют способность агонистического лиганда вызывать полный максимальный рецепторный ответ ткани или системы-мишени. Частичный агонизм означает способность лиганда к получению субмаксимальной амплитуды биологического ответа. Предполагается, что частичный агонист вызывает агонизм в низких дозах в системах, где отсутствуют другие агонисты, тогда как в присутствии полного агониста он действует как конкурентный антагонист, поскольку конкурирует с полным агонистом за связывание с рецептором. Тем не менее, частичный агонист с высокой внутренней активностью может вызывать дополнительную активацию рецепторов из-за сберегающего действия рецепторов в отношении друг друга даже в присутствии высоких уровней эндогенного агониста. Кроме того, считается, что очень низкая внутренняя активность соединений не обеспечит достижение пороговых уровней ответа, а просто позволит занять рецептор, что приведет к получению окончательного антагонистического эффекта. Антагонист представляет собой лиганд, который обладает сродством, но не имеет внутренней активности в отношении родственного рецептора, а связывание нарушает взаимодействие и ослабляет биологический эффект агониста. В зависимости от типа молекулярного связывания с рецептором и конкуренции с эндогенным лигандом рецептора антагонисты могут быть конкурентными и неконкурентными, обратимыми и необратимыми. Наконец, лиганды с отрицательной внутренней активностью были названы обратными агонистами. В зависимости от состояния активности рецептора эти соединения могут демонстрировать эффекты, сходные с эффектами антагонистов.
Кроме того, считается, что очень низкая внутренняя активность соединений не обеспечит достижение пороговых уровней ответа, а просто позволит занять рецептор, что приведет к получению окончательного антагонистического эффекта. Антагонист представляет собой лиганд, который обладает сродством, но не имеет внутренней активности в отношении родственного рецептора, а связывание нарушает взаимодействие и ослабляет биологический эффект агониста. В зависимости от типа молекулярного связывания с рецептором и конкуренции с эндогенным лигандом рецептора антагонисты могут быть конкурентными и неконкурентными, обратимыми и необратимыми. Наконец, лиганды с отрицательной внутренней активностью были названы обратными агонистами. В зависимости от состояния активности рецептора эти соединения могут демонстрировать эффекты, сходные с эффектами антагонистов.
Однако картина усложняется рядом дополнительных фармакологических свойств, которые также следует принимать во внимание. Наблюдаемая удельная активность лекарственного средства (т. е. биологический эффект, который внешний лиганд будет производить в системе) представляет собой сложную функцию его сродства и внутренней активности и пропорциональна их значениям. Таким образом, препарат с высокой удельной активностью может проявлять высокое сродство, или высокую внутреннюю активность, или оба свойства, что также зависит от чувствительности системы. В тканях с низкой чувствительностью препараты, которые проявляют высокую удельную активность из-за высокой внутренней активности, производят более устойчивые эффекты, в то время как агонисты с высокой активностью на фоне высокого сродства будут вести себя как частичные агонисты или антагонисты. Другой первичной молекулярной особенностью препарата является его селективность, которая напрямую зависит от концентрации молекулы с определенным сродством и внутренней активностью, вызывающей эффекты только в конкретной системе-мишени (системе направленной доставки препарата). Более того, механизм взаимодействия лиганда представлен его способностью связывать сайт эндогенного лиганда (ортостерическое взаимодействие) или свой собственный сайт на рецепторе, приводя к изменению конформации и, следовательно, активности рецептора (аллостерическое взаимодействие).
е. биологический эффект, который внешний лиганд будет производить в системе) представляет собой сложную функцию его сродства и внутренней активности и пропорциональна их значениям. Таким образом, препарат с высокой удельной активностью может проявлять высокое сродство, или высокую внутреннюю активность, или оба свойства, что также зависит от чувствительности системы. В тканях с низкой чувствительностью препараты, которые проявляют высокую удельную активность из-за высокой внутренней активности, производят более устойчивые эффекты, в то время как агонисты с высокой активностью на фоне высокого сродства будут вести себя как частичные агонисты или антагонисты. Другой первичной молекулярной особенностью препарата является его селективность, которая напрямую зависит от концентрации молекулы с определенным сродством и внутренней активностью, вызывающей эффекты только в конкретной системе-мишени (системе направленной доставки препарата). Более того, механизм взаимодействия лиганда представлен его способностью связывать сайт эндогенного лиганда (ортостерическое взаимодействие) или свой собственный сайт на рецепторе, приводя к изменению конформации и, следовательно, активности рецептора (аллостерическое взаимодействие). Наконец, время удержания представляет собой показатель охвата целевой кинетики, который зависит как от фармакокинетики соединения in vivo, так и от скорости диссоциации рецептора.
Наконец, время удержания представляет собой показатель охвата целевой кинетики, который зависит как от фармакокинетики соединения in vivo, так и от скорости диссоциации рецептора.
Понятия агонизма, антагонизма, а также обратного агонизма считаются основой понимания лиганд-рецепторного взаимодействия. Эти свойства молекулы считаются в одинаковой степени влияющими на все вторичные каскады мессенджеров в зависимости от рецептора или системы, на которые воздействует лиганд. Внутренняя активность рассматривается как параметр, не зависящий от системы. Это означает, что полный агонист будет постоянно вызывать полный рецепторный ответ независимо от ткани и условий, в которых происходит экспрессия рецептора. Кроме того, предполагается, что полный агонист активирует все сигнальные пути, управляемые его родственным рецептором, а антагонист блокирует все нисходящие сигнальные пути, связанные с антагонизированным рецептором.
В последнее время эти понятия были пересмотрены благодаря появлению нового понятия «функциональная селективность», которое предполагает, что препарат нелегко классифицировать как агонист или антагонист, поскольку он может вызывать комбинацию классических эффектов посредством активации или ингибирования только одного или ограниченного числа путей передачи сигналов, связанных с рецептором-мишенью. Этот эффект может быть обусловлен индукцией уникальных лиганд-специфичных конформаций рецепторного комплекса-мишени, что приводит к дифференциальной активации одного или нескольких путей передачи нисходящих сигналов рецептора. Подобные молекулярные эффекты могут зависеть от сигнального аппарата, связанного с рецептором, и, следовательно, от типа и локализации клеток, экспрессирующих рецептор-мишень.
Этот эффект может быть обусловлен индукцией уникальных лиганд-специфичных конформаций рецепторного комплекса-мишени, что приводит к дифференциальной активации одного или нескольких путей передачи нисходящих сигналов рецептора. Подобные молекулярные эффекты могут зависеть от сигнального аппарата, связанного с рецептором, и, следовательно, от типа и локализации клеток, экспрессирующих рецептор-мишень.
Фармакодинамические свойства арипипразола
вверх
Рецепторный профиль
В таблице 1 показано сродство арипипразола к ряду соответствующих рецепторов центральной нервной системы. Среди дофаминовых рецепторов арипипразол отличает высокое сродство к подтипам D2R и D3R, тогда как в отношении подтипов D1 (D1R), D4 (D4R) и D5 (D5R) оно ограничено или незначительно. Среди серотонинергических рецепторов арипипразол имеет очень высокое и высокое сродство к рецепторам 5-HT1A (5-HT1AR), 5-HT2A (5-HT2AR), 5-HT2B (5-HT2BR) и 5-HT7 (5HT7R). Сродство является умеренным к рецепторам 5-HT1D (5HT1DR) и 5-HT2C (5-HT2CR), ограниченным – к рецепторам 5-HT1B (5-HT1BR), 5-HT3 (5-HT3R) и 5-HT6 (5HT6R) и незначительным – к рецепторам 5-HT1E (5-HT1ER), 5-HT5 (5-HT5R) и транспортерам серотонина.
Сродство является умеренным к рецепторам 5-HT1D (5HT1DR) и 5-HT2C (5-HT2CR), ограниченным – к рецепторам 5-HT1B (5-HT1BR), 5-HT3 (5-HT3R) и 5-HT6 (5HT6R) и незначительным – к рецепторам 5-HT1E (5-HT1ER), 5-HT5 (5-HT5R) и транспортерам серотонина.
Таблица 1. Рецепторный профиль и функциональное действие арипипразола
|
Тип рецептора |
Константа сродства (Ki) |
Типология |
Биологическое действие против агонистического |
Экспериментальная система |
Клетки/ткани, экспрессирующие рецепторы-мишени |
Источник |
|
Дофамин |
||||||
|
D1 |
− |
Крыса |
Клетки CHO |
Lawler et al. |
||
|
− |
Человек |
Shapiro et al., 2003 |
||||
|
D2 |
++ |
Крыса |
Полосатое тело крыс |
Lawler et al., 1999 |
||
|
+++ |
Крыса (D2S) |
Антагонизм |
Ингибирование продукции цАМФ, индуцированной форсколином |
CHO |
||
|
+++ |
Крыса (D2L) |
CHO |
||||
|
++ |
Крыса (D2L) |
Частичный агонизм |
Стимулирование накопления цАМФ, стимулируемое изопротеренолом |
Глиома C6 |
||
|
+++ |
Человек |
Антагонизм |
Активность ГТФазы, индуцированная квинпиролом |
Стриарные мембраны головного мозга крысы |
Inoue et al. |
|
|
Частичный агонизм |
Ингибирование продукции цАМФ, индуцированной форсколином |
CHO |
Burris et al., 2002 |
|||
|
Антагонизм |
Модуляция токов GIRK |
MES-23.5 |
Shapiro et al., 2003 |
|||
|
Антагонизм |
Связывание GTPyS |
CHO |
||||
|
Частичный агонизм |
Ингибирование высвобождения пролактина, индуцированного форсколином Ингибирование продукции цАМФ, индуцированной форсколином |
GFh5C1 |
Aihara et al. |
|||
|
Частичный агонизм |
Ингибирование продукции цАМФ, индуцированной форсколином |
CHO |
Tadori et al., 2005 |
|||
|
Частичный агонизм |
Ингибирование фосфорилирования ERK, индуцированного дофамином |
CHO |
Bruins Slot et al., 2006 |
|||
|
Частичный агонизм |
D2-опосредованное фосфорилирование MAPK D2-опосредованное потенцирование высвобождения арахидоновой кислоты |
CHO |
Urban et al. |
|||
|
Агонизм |
Ингибирование продукции цАМФ, индуцированной форсколином |
CHO (hD2SR с низкой плотностью) |
Tadori et al., 2011 |
|||
|
Частичный агонизм |
CHO (hD2SR с высокой плотностью) |
|||||
|
Антагонизм |
CHO (hD2LR с низкой плотностью) |
|||||
|
Частичный агонизм |
CHO (hD2LR с высокой плотностью) |
|||||
|
D3 |
++ |
Крыса |
Lawler et al. |
|||
|
++ |
Крыса |
Глиома C6 |
||||
|
++ |
Человек |
Частичный агонизм |
Ингибирование продукции цАМФ, индуцированной форсколином |
CHO |
Tadori et al., 2008 |
|
|
Частичный агонизм |
Ингибирование продукции цАМФ, индуцированной форсколином |
CHO |
Tadori et al. |
|||
|
D4 |
+/− |
Крыса |
CHO |
Lawler et al., 1999 |
||
|
+/− |
Крыса |
Частичный агонизм |
Модуляция токов GIRK |
Ооциты африканской шпорцевой лягушки |
Newmann-Tancredi et al., 2008 |
|
|
D5 |
− |
Крыса |
CHO |
Lawler et al. |
||
|
− |
Человек |
Shapiro et al., 2003 |
||||
|
DAT |
− |
Человек |
||||
|
Серотонин |
||||||
|
5-HT1A |
++ |
Человек |
Частичный агонизм |
Ингибирование продукции цАМФ, индуцированной форсколином |
CHO |
Shapiro et al. |
|
Частичный агонизм |
Связывание GTPyS |
CHO |
Jordan et al., 2002 |
|||
|
Частичный агонизм |
Связывание GTPyS |
Гипокампальные мембраны крысы |
Stark et al., 2007 |
|||
|
Агонизм |
Частота вспышек активности |
5-HT дорсального ядра шва |
Stark et al. |
|||
|
5-HT1B |
+/− |
Человек |
Shapiro et al., 2003 |
|||
|
5-HT1D |
+ |
Человек |
Shapiro et al., 2003 |
|||
|
5-HT1E |
− |
Человек |
Shapiro et al. |
|||
|
5-HT2A |
++ |
Человек |
Частичный агонизм |
Стимулирование гидролиза ИП |
Клетки глиомы C6, экспрессирующие эндогенные 5-HT2AR GF62 |
Shapiro et al., 2003 |
|
5-HT2B |
+++ |
Человек |
Обратный агонизм |
Стимулирование гидролиза ИП |
HEK-293 |
Shapiro et al. |
|
5-HT2C |
+ |
Человек |
Частичный агонизм Агонизм |
Стимулирование гидролиза ИП Стимулирование гидролиза ИП |
PO1C COS-7 |
Shapiro et al., 2003 |
|
5-HT3 |
+/− |
Крыса |
Shapiro et al., 2003 |
|||
|
5-HT5A |
− |
Человек |
Shapiro et al. |
|||
|
5-HT6 |
+/− |
Крыса |
HEK-293 |
Lawler et al., 1999 |
||
|
+/− |
Человек |
Антагонизм |
Стимулирование продукции цАМФ |
HEK-293 |
Shapiro et al., 2003 |
|
|
5-HT7 |
+ |
Крыса |
HEK-293 |
Lawler et al. |
||
|
++ |
Человек |
Частичный агонизм |
Стимулирование продукции цАМФ |
HEK-293 |
Shapiro et al., 2003 |
|
|
SERT |
− |
Человек |
Shapiro et al., 2003 |
|||
|
Норадреналин |
||||||
|
α1A |
+ |
Человек |
Антагонизм |
Shapiro et al. |
||
|
α1B |
+ |
Человек |
Антагонизм |
|||
|
α2A |
+ |
Человек |
Антагонизм |
|||
|
α2B |
+/− |
Человек |
||||
|
α2C |
+ |
Человек |
Антагонизм |
|||
|
β1 |
+/− |
Человек |
||||
|
β2 |
+/− |
Человек |
||||
|
NET |
− |
Человек |
||||
|
Гистамин |
||||||
|
h2 |
+ |
Человек |
Антагонизм |
Shapiro et al. |
||
|
h3 |
− |
Человек |
||||
|
h4 |
+/− |
Морская свинка |
||||
|
h5 |
− |
Человек |
||||
|
Ацетилхолин |
||||||
|
M1 |
− |
Человек |
Shapiro et al. |
|||
|
M2 |
− |
Человек |
||||
|
M3 |
− |
Человек |
||||
|
M4 |
− |
Человек |
||||
|
M5 |
− |
Человек |
||||
|
ГАМК |
||||||
|
ГАМК-A |
− |
Крыса |
Shapiro et al. |
|||
|
ГАМК-B |
− |
Крыса |
||||
|
Глутамат |
||||||
|
NMDA |
− |
Крыса |
Shapiro et al., 2003 |
|||
|
Опиат |
||||||
|
Κ |
− |
Человек |
Shapiro et al. |
|||
|
Μ |
− |
Человек |
||||
|
Δ |
− |
Человек |
||||
|
Примечания: там, где возможно, приведены результаты функциональных исследований по оценке внутренней активности арипипразола на рецепторах-мишенях по сравнению с агонистом; цАМФ – циклический аденозинмонофосфат; CHO (Chinese hamster ovary) – клетки яичников китайского хомячка; GIRK (G-protein-regulated inwardly rectifying potassium channel) – G-белок связанные калиевые каналы внутреннего выпрямления; GTP – γ-трифосфат; hD2LR – длинный D2-рецептор человека; hD2SR – короткий D2-рецептор человека; HEK (human embryonic kidney) – почка эмбриона человека; MAPK (Mitogen-Activated Protein Kinase) – митоген-активированная протеинкиназа;
«+++» означает ≤ 1 – очень высокое сродство; «++» означает ≤ 10 – высокое сродство; «+» означает ≤ 100 – умеренное сродство; «+/−» означает ≤ 1000 – ограниченное сродство; «−» означает > 1000 – незначительное сродство. |
||||||
 , 1999
, 1999 , 1997
, 1997 , 2004
, 2004 , 2007
, 2007 , 1999
, 1999 , 2011
, 2011 , 1999
, 1999 , 2003
, 2003 , 2007
, 2007 , 2003
, 2003 , 2003
, 2003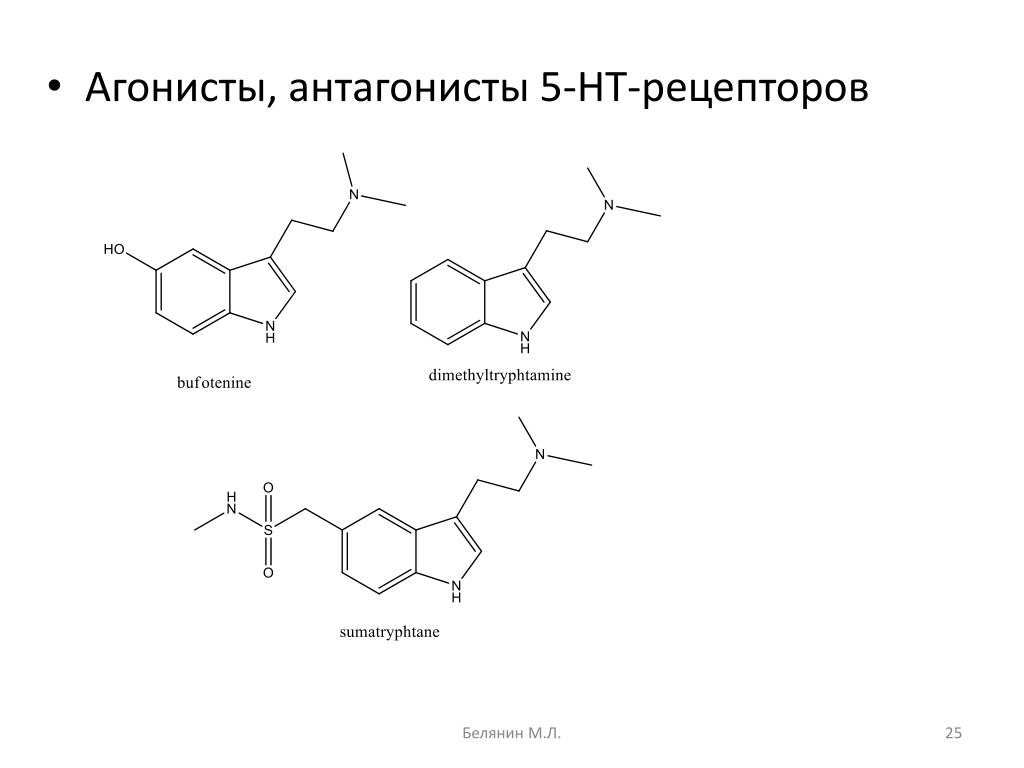 , 2003
, 2003 , 1999
, 1999 , 2003
, 2003 , 2003
, 2003 , 2003
, 2003 , 2003
, 2003 , 2003
, 2003
Среди других нейротрансмиттерных систем арипипразол обладает умеренным сродством к адренергическим рецепторам α1A, α1B, α2A и α2C, а также к гистаминергическим рецепторам h2 (h2R) (см. табл. 1). Соединение также обладает ограниченным сродством к α2В, β1, β2 и Н3-рецепторам. Незначительным является к холинергическим, ГАМК-эргическим (γ-аминомасляная кислота), глутаматергическим и опиоидергическим рецепторам.
Молекулярное действие арипипразола на D2-рецепторы дофамина (D2R): от частичного агонизма к функциональной селективности
Арипипразол имеет высокое сродство к D2R, что является одним из самых высоких показателей в отношении этого рецептора для антипсихотиков (константа сродства Ki = 0,34 нМ). Несмотря на низкую частоту возникновения острых экстрапирамидных побочных эффектов, было доказано, что в терапевтических дозах соединение занимает до 95% D2R в стриатуме, что значительно превышает пороговое значение, необходимое антипсихотическим препаратам для их инициирования. С другой стороны, препарат стимулирует D2R только в субмаксимальной фракции стимулирования, вызываемого эндогенным лигандом, то есть дофамином. Поэтому внутренняя активность арипипразола в отношении D2R может оказаться ниже, чем таковая дофамина.
С другой стороны, препарат стимулирует D2R только в субмаксимальной фракции стимулирования, вызываемого эндогенным лигандом, то есть дофамином. Поэтому внутренняя активность арипипразола в отношении D2R может оказаться ниже, чем таковая дофамина.
Кроме того, арипипразол, в отличие от дофамина, не опосредовал значительную интернализацию D2R. Эти данные показывают, что арипипразол ведет себя как лиганд со смещенной активностью в отношении рецепторов D2R/D3R, проявляя поочередно частичные агонистические или антагонистические свойства при взаимодействии с ними.
Основная концепция, поддерживаемая на протяжении многих лет, заключается в том, что арипипразол, в отличие от других антипсихотиков, может оказывать дифференциальное действие на D2R и выступать в качестве преобладающего антагониста, когда синаптические концентрации дофамина повышаются, или действовать как преобладающий частичный агонист D2R, когда концентрации дофамина низки. Было высказано предположение, что сочетание тесного связывания с D2R и частичного агонизма к этому рецептору может объяснить благоприятный профиль терапевтической эффективности / побочных эффектов арипипразола.
В частности, арипипразол может подавлять высвобождение фазового дофамина вследствие вспышки активности дофаминергических нейронов, что может объяснить его терапевтическую эффективность, тогда как тоническое высвобождение дофамина может относительно сохраняться из-за частичного агонистического действия соединения. Эта особенность отличает арипипразол от антипсихотиков с полным антагонистическим действием и плотным связыванием на D2R, которые подавляют как тоническое, так и фазовое высвобождение дофамина, что может служить объяснением склонности этих соединений к индуцированию экстрапирамидных побочных эффектов.
Совсем недавно эта концепция была надлежащим образом обоснована свойствами арипипразола в отношении D2R-подобных ауторецепторов путем обнаружения изменений синтеза дофамина в стриарной ткани головного мозга крыс, инкубированной ex vivo.
Однако вывод заключается о том, что арипипразол вызывает множественные D2R-опосредованные функциональные эффекты, охватывающие весь спектр фармакологических признаков, и это позволило прогрессивно изменить описание механизма действия этого препарата, обосновывая гипотезу функциональной селективной модуляции D2R-зависимых специфических нисходящих сигнальных путей. Сообщалось, что предполагаемые частичные агонисты могут проявлять лиганд-зависимую модуляцию микропереключателей, относящуюся к активации рецептора и субмаксимальному связыванию с внутриклеточными эффекторами. Кроме того, эти агенты могут регулировать лиганд-специфичные конформации в рецептор-специфических эпитопах, включая области связывания рецептора с G-белком. В гомологической модели активного состояния комплекса D2R-Gαi-белок арипипразол индуцировал различные конформации необходимых структурных мотивов, включая области внеклеточной петли, связывающий «карман» и внутриклеточные домены связывания G-белка. Эти лиганд-специфичные конформационные изменения имеют функциональные последствия. В системе экспрессии ооцитов арипипразол немного уменьшал токи через G-белок связанный калиевый канал внутреннего выпрямления (GIRK), ко-экспрессируемый hD2LR, тогда как действие многих антипсихотиков такие токи полностью ликвидировало. Эти наблюдения скорее согласуются с моделью «функциональной селективности», указывая на то, что препарат может вызывать множественные функциональные эффекты даже при взаимодействии с одной изоформой рецептора.
Сообщалось, что предполагаемые частичные агонисты могут проявлять лиганд-зависимую модуляцию микропереключателей, относящуюся к активации рецептора и субмаксимальному связыванию с внутриклеточными эффекторами. Кроме того, эти агенты могут регулировать лиганд-специфичные конформации в рецептор-специфических эпитопах, включая области связывания рецептора с G-белком. В гомологической модели активного состояния комплекса D2R-Gαi-белок арипипразол индуцировал различные конформации необходимых структурных мотивов, включая области внеклеточной петли, связывающий «карман» и внутриклеточные домены связывания G-белка. Эти лиганд-специфичные конформационные изменения имеют функциональные последствия. В системе экспрессии ооцитов арипипразол немного уменьшал токи через G-белок связанный калиевый канал внутреннего выпрямления (GIRK), ко-экспрессируемый hD2LR, тогда как действие многих антипсихотиков такие токи полностью ликвидировало. Эти наблюдения скорее согласуются с моделью «функциональной селективности», указывая на то, что препарат может вызывать множественные функциональные эффекты даже при взаимодействии с одной изоформой рецептора.
Один гипотетический механизм функциональной селективности (который может в надлежащей степени соответствовать экспериментальным данным в отношении биологического действия арипипразола) состоит в том, что таргетный препарат может инициировать различные изменения конформации по сравнению с эндогенным лигандом при связывании с комплексом рецептор-белок G. Такой функциональный таргетинг обеспечивает усовершенствование эффектов лекарственного средства, которые не могут быть достигнуты путем простого нацеливания на конкретные рецепторные изоформы.
Переходя к клиническому применению, следует отметить, что в настоящее время арипипразол, по сравнению с частичными агонистами с потенциальной антипсихотической активностью, является единственным препаратом, позволившим достичь успеха на всех клинических этапах, необходимых для утверждения и использования препарата у пациентов, тогда как попытки использовать другие соединения потерпели неудачу вследствие прогрессирующей потери эффективности (как в случае с прекламолом (3PPP)) или возникновения нежелательных явлений (как в случае с OPC-4392, предшественником арипипразола).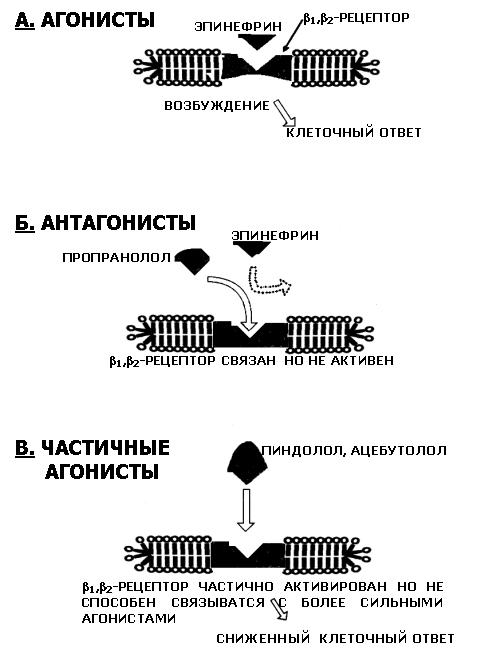
Рисунок. Постсинаптическая функциональная селективность арипипразола
Рисунок. Постсинаптическая функциональная селективность арипипразола
Таким образом, арипипразол может вести себя как функционально-селективный агент, причем его внутренняя активность сильно варьирует и зависит от экспериментальной парадигмы, а также от сигнальной среды D2R. Схематическая модель возможных механизмов, объясняющих постсинаптическую функциональную селективность арипипразола, представлена на рисунке. Предполагается, что арипипразол обусловливает функциональную множественность на постсинаптических нисходящих путях D2-рецепторов дофамина. Одним из механизмов функциональной селективности может быть предпочтительное связывание с различными конформациями D2-рецепторов, которые, как было показано, активируют дифференциальные трансдукционные пути в соответствии с подтипами нейронов, в которых они экспрессируются. Постсинаптические поддерживающие белки/адаптеры и эффекторы могут дифференцированно воздействовать на каждый путь, связанный с конформацией D2-рецепторов дофамина, селективно активируемый арипипразолом.
Функциональная значимость взаимодействий с другими рецепторами, помимо D2R
В многочисленных анализах in vitro арипипразол показал себя как частичный агонист в отношении 5-HT1A-рецепторов. Ингибирование активности нейронов серотонина арипипразолом опосредовано его агонистическим действием на соматодендритные 5-HT1AR-ауторецепторы, расположенные на серотонинергических нейронах дорсального ядра шва. Полная агонистическая активность может быть объяснена большим запасом 5-HT1AR в этих ядрах и иметь отношение к общему фармакологическому действию арипипразола. Серотонинергические нейроны дорсального ядра шва обеспечивают основные афферентные проекции на лобную кору. Действие агониста на соматодендритные 5-HT1AR снижает высвобождение серотонина в коре. Это в свою очередь стимулирует высвобождение в ней дофамина, что может быть полезным в лечении негативных и когнитивных симптомов шизофрении. Стоит отметить, что ухудшение функции распознавания, вызванное повторным применением фенциклидина, уменьшилось под действием арипипразола, благотворные эффекты которого в свою очередь были отменены совместным применением антагониста D1R и антагониста 5-HT1AR.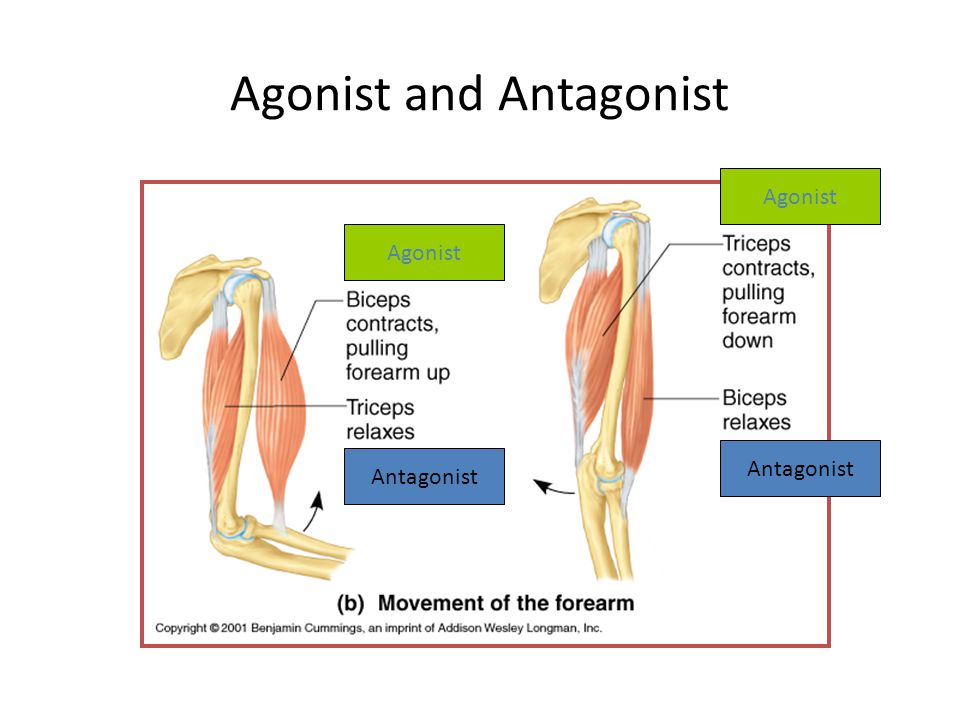
Кроме того, арипипразол уменьшил проявления нарушений социального поведения, вызванных повторным применением фенциклидина, – эффект, который был предотвращен предварительным лечением с применением 5-HT1AR антагониста. Более того, посредством воздействия на 5-HT1AR арипипразол подавляет поведение «закапывания шариков» (marble-burying behavior) в соответствующем тесте, животной модели обсессивно-компульсивного расстройства. Низкие дозы системного арипипразола (< 1 мг/кг) также увеличивали уровни внеклеточного дофамина в коре. С другой стороны, высокие дозы арипипразола (10-40 мг/кг) при системной терапии снижали уровни дофамина. Этот двухфазный эффект может зависеть от распространенности агонистических 5-HT1AR-опосредованных эффектов в низких дозах и распространенности ингибирования активности мезокортикальных дофаминергических нейронов в высоких дозах.
С трансляционной точки зрения действие арипипразола на 5-HT1AR ассоциировалось с потенциальной модуляцией когнитивных эффектов.
Арипипразол обладает активностью частичного агониста/антагониста в отношении 5-HT1AR. Что касается других серотонинергических рецепторов, арипипразол вел себя как обратный агонист 5-HT2BR, как частичный агонист с высокой внутренней активностью в отношении 5-HT2CR, как антагонист 5-HT6R и как слабый частичный агонист 5-HT7R. Эффекты в отношении 5-HT2CR сильно зависели от рассматриваемой системы. Существуют данные о том, что арипипразол может вести себя как полный агонист 5-HT2CR (когда серотониновый тонус является низким, а рецепторный резерв – высоким) и как антагонист (в случаях высокого серотонинового тонуса / низкого рецепторного резерва). Агонизм в отношении 5-HT2CR считается полезным для снижения аппетита и предотвращения увеличения массы тела, индуцированного приемом препарата.
С учетом этих факторов, когда арипипразол применили в комбинации с серотонинергическими антидепрессантами с высокой активностью, было обнаружено статистически значимое увеличение массы тела, что, возможно, было обусловлено повышением серотонинового тонуса под действием последних соединений. Интересно, что тот же эффект не был отмечен, когда арипипразол комбинировали с серотонинергическими антидепрессантами с низкой активностью.
Интересно, что тот же эффект не был отмечен, когда арипипразол комбинировали с серотонинергическими антидепрессантами с низкой активностью.
Наконец, актуальной проблемой трансляционной медицины, обусловленной сложностью рецепторного профиля арипипразола, является совместное назначение этого препарата с другими психотропными соединениями. Такие комбинации часто используются в клинической практике для решения многих аспектов тяжелых психических заболеваний (таких как шизофрения и биполярные расстройства) или противодействия развитию шизофрении, резистентной к антипсихотикам. Например, комбинация клозапина и арипипразола может привести к большему антагонизму в отношении D2R в мезолимбических путях и, кроме того, может сочетаться с антагонизмом в отношении D2R и D4R.
Влияние на транскрипцию молекул, имеющих отношение к патофизиологии психоза
Арипипразол-опосредованные изменения транскриптома нейронов в соответствующих биологических функциях изучали с помощью анализа экспрессии генов. Эти исследования также обнаружили существенные различия изменений транскриптома, обусловленных арипипразолом и другими антипсихотиками, подтвердившие мнение о том, что арипипразол воздействует на специфические по сравнению с традиционными антипсихотическими средствами нейробиологические пути. В таблице 2 приведены наиболее актуальные исследования арипипразол-опосредованной экспрессии генов в центральной нервной системе.
Эти исследования также обнаружили существенные различия изменений транскриптома, обусловленных арипипразолом и другими антипсихотиками, подтвердившие мнение о том, что арипипразол воздействует на специфические по сравнению с традиционными антипсихотическими средствами нейробиологические пути. В таблице 2 приведены наиболее актуальные исследования арипипразол-опосредованной экспрессии генов в центральной нервной системе.
Таблица 2.
Эффекты арипипразола на экспрессию генов в центральной нервной системе|
Ген |
Уровни экспрессии |
Экспериментальная парадигма |
Штамм |
Область головного мозга |
Метод |
Источник |
|
DRD2 |
Не затронут |
12 или 100 мг/кг п/о 21 день |
Крысы линии Вистар |
Стриатум |
Анализ с помощью защиты от рибонуклеазы |
Inoue et al. |
|
↓ |
24 мг/кг п/о 21 день |
Крысы линии Вистар |
Гипофиз |
Анализ с помощью защиты от рибонуклеазы |
Inoue et al., 1998 |
|
|
10 мг/кг в/б 21 день |
RT-PCR |
Luoni et al. |
||||
|
Homer1a |
↑ |
12 мг/кг в/б Острое применение |
Крысы линии Спрег-Доули |
CP |
ISHH |
Tomasetti et al., 2007 |
|
↑ |
30 мг/кг в/б Острое применение |
FrC ACC Оболочка прилежащего ядра |
||||
|
↑ |
12 мг/кг в/б 21 день |
FrC ACC Латеральная CP |
||||
|
Arc |
↑ |
10 мг/кг IP 21 день |
Крысы линии Спрег-Доули |
Гиппокамп PFC Стриатум |
qRT-PCR |
Luoni et al. |
|
10 мг/кг в/б 21 день + острый стресс |
Гиппокамп PFC |
|||||
|
↓ |
0,08 мг/кг п/к Острое применение |
Крысы линии Спрег-Доули |
Стриатум |
RT-PCR |
Waters et al., 2014 |
|
|
BDNF |
↓ |
10 мг/кг в/б 21 день |
Крысы линии Спрег-Доули |
Гиппокамп |
qRT-PCR |
Luoni et al. |
|
↑ |
10 мг/кг в/б 21 день + острый стресс |
PFC |
||||
|
Примечания: ACC – передняя поясная кора; CP – дорсолатеральный отдел стриатума; FrC – лобная кора; в/б – внутрибрюшинно; ISHH – гистохимическая гибридизация in situ; PFC – префронтальная кора; п/о – перорально; qRT-PCR – количественный анализ с использованием полимеразной цепной реакции в режиме реального времени, п/к – подкожно. |
||||||
 , 1997
, 1997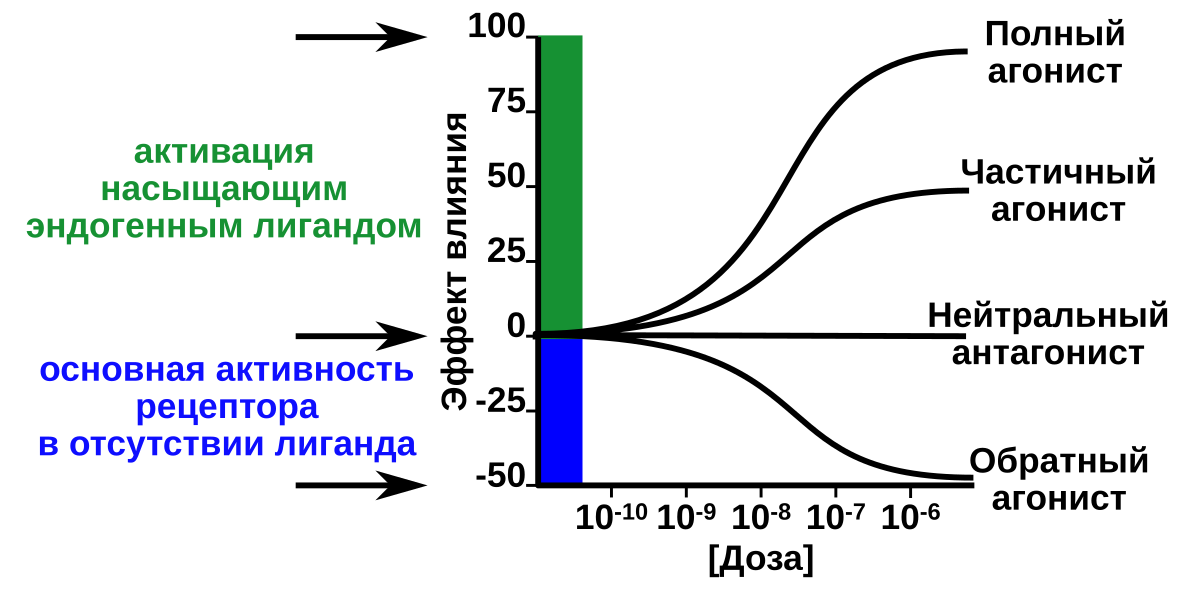 , 2014
, 2014 , 2014
, 2014 , 2014
, 2014
Было обнаружено, что арипипразол выборочно модулирует экспрессию генов, вовлеченных в дофаминергическую передачу сигналов в областях головного мозга, принадлежащих к мезолимбическим и нигростриарным дофаминовым путям. Действительно, 12-недельное лечение с применением пероральной дозы арипипразола 2,25 мг/кг значительно увеличивает экспрессию матричной РНК (мРНК) D2R и уменьшает экспрессию мРНК тирозингидроксилазы в вентральной области покрышки среднего мозга крысы на фоне отсутствия эффектов в черном веществе или прилежащем ядре. И наоборот, было отмечено, что галоперидол увеличивает плотность связывания D2R в прилежащем ядре и дорсолатеральном отделе стриатума, а также уменьшает связывание дофаминового транспортера (DAT) в этих двух областях и в области покрышки среднего мозга. Оланзапин может увеличивать содержание мРНК D2R в области покрышки среднего мозга и связывание DAT в прилежащем ядре. Таким образом, эти результаты показывают, что арипипразол оказывает более избирательное воздействие на мезолимбический дофаминергический путь, чем галоперидол и оланзапин.
Действительно, 12-недельное лечение с применением пероральной дозы арипипразола 2,25 мг/кг значительно увеличивает экспрессию матричной РНК (мРНК) D2R и уменьшает экспрессию мРНК тирозингидроксилазы в вентральной области покрышки среднего мозга крысы на фоне отсутствия эффектов в черном веществе или прилежащем ядре. И наоборот, было отмечено, что галоперидол увеличивает плотность связывания D2R в прилежащем ядре и дорсолатеральном отделе стриатума, а также уменьшает связывание дофаминового транспортера (DAT) в этих двух областях и в области покрышки среднего мозга. Оланзапин может увеличивать содержание мРНК D2R в области покрышки среднего мозга и связывание DAT в прилежащем ядре. Таким образом, эти результаты показывают, что арипипразол оказывает более избирательное воздействие на мезолимбический дофаминергический путь, чем галоперидол и оланзапин.
Более ранние исследования также продемонстрировали, что арипипразол может значительно снизить экспрессию генов D2R в гипофизе крыс, тогда как галоперидол способствует увеличению содержания мРНК как D2R, так и пролактина, что на молекулярном уровне объясняет эффективность арипипразола для преодоления гиперпролактинемии, индуцированной антипсихотиками.
Кроме того, было установлено, что арипипразол воздействует на экспрессию гена, связанную с системами нейротрансмиттеров, кроме дофамина. Применение арипипразола в течение 1 недели ассоциировалось с увеличением связывания 5-HT1AR в гиппокампе, хотя без увеличения экспрессии мРНК 5-HT1AR. Однако эти эффекты не сохраняются после длительного применения арипипразола. Кроме того, было продемонстрировано, что арипипразол, в отличие от оланзапина, не влияет на экспрессию мРНК h2R в дугообразном гипоталамическом ядре, напрямую коррелирующую с потреблением пищи и увеличением массы тела, связанным с приемом антипсихотиков. Есть сообщения о повышенной экспрессии мРНК мускариновых M1-рецепторов (M1R) в гиппокампе и прилежащем ядре при длительном применении арипипразола у крыс без влияния на экспрессию нигростриатных M1R, которую могут модулировать оланзапин и галоперидол.
Что касается ГАМК-эргической нейротрансмиссии, было доказано, что длительный прием арипипразола уменьшает экспрессию мРНК гена декарбоксилазы глутаминовой кислоты (GAD67), который кодирует фермент, играющий главную роль в синтезе ГАМК, в дорсолатеральном отделе стриатума, префронтальной коре и соматосенсорной коре. В то же время экспрессия гена GAD67 может индуцироваться в гиппокампе, гипоталамусе, таламусе и миндалевидном теле. Были установлены некоторые другие значительные изменения в экспрессии мРНК генов, кодирующих везикулярный транспортер ГАМК (Slc32a1) и трансмембранный транспортер Slc6a1 и Slc6a11.
В то же время экспрессия гена GAD67 может индуцироваться в гиппокампе, гипоталамусе, таламусе и миндалевидном теле. Были установлены некоторые другие значительные изменения в экспрессии мРНК генов, кодирующих везикулярный транспортер ГАМК (Slc32a1) и трансмембранный транспортер Slc6a1 и Slc6a11.
Длительное применение арипипразола также может индуцировать экспрессию мРНК субъединиц NR1, NR2A и NR2C рецептора N-метил-D-аспартата (NMDA) с одновременным снижением содержания субъединицы NR2B. Это способствует сдвигу соотношения NR2A/NR2B субъединиц NMDA к типу, соответствующему взрослым особям, что ведет к расширению функции и экспрессии NMDA. Подтвердилось, что арипипразол влияет на экспрессию генов, кодирующих транспортеры глутамата. В частности, арипипразол может подавлять экспрессию мРНК генов глиальных транспортеров возбуждающих аминокислот EAAT1 и EAAT2 и нейронального транспортера EAAT3 в гиппокампальных субрегионах и EAAT4 в лобной коре, одновременно увеличивая экспрессию пресинаптического везикулярного глутаматного транспортера VGluT1 в гиппокампе, что способствует усилению глутаматной нейротрансмиссии в этих областях.
В целом вышеупомянутые исследования экспрессии генов показали, что арипипразол может оказывать комплексные зависимые от дозы и времени эффекты на множественные нейротрансмиттерные системы, не включенные в рецепторный профиль арипипразола (например, глутаматергическую) и не являющиеся непосредственной мишенью соединения. Результаты этих исследований показывают, что арипипразол индуцирует широкий и еще не полностью описанный диапазон молекулярных эффектов в центральной нервной системе, и позволяют предположить, что они могут быть весьма полезными для объяснения глобальной терапевтической эффективности и долгосрочных нейробиологических связей за пределами его рецепторного профиля.
Анализ экспрессии генов также продемонстрировал влияние арипипразола на молекулы, вовлеченные в синаптическую пластичность. Эксперименты с применением метода полимеразной цепной реакции в режиме реального времени (RT-PCR) показали, что арипипразол может снижать экспрессию мРНК регулируемого активностью ассоциированного с цитоскелетом белка (activity-regulated cytoskeleton-associated protein) / регулируемого активностью гена 3. 1 (activity-regulated gene 3.1) (Arc – немедленно-ранний ген, который индуцируется синаптической активностью и регулирует реорганизацию цитоскелета в дендритных шипиках) в стриатуме крысы, тогда как галоперидол может увеличивать ее.
1 (activity-regulated gene 3.1) (Arc – немедленно-ранний ген, который индуцируется синаптической активностью и регулирует реорганизацию цитоскелета в дендритных шипиках) в стриатуме крысы, тогда как галоперидол может увеличивать ее.
Последние данные свидетельствуют о том, что арипипразол также может непрямо модулировать глутаматергическую сигнализацию в областях головного мозга, ключевых для антипсихотической терапии, таких как латеральный отдел стриатума и передняя поясная кора. Одним из белков, связанных с аномальной синаптической пластичностью, который, как считается, участвует в развитии шизофрении, является нейрегулин 1 (NRG1). Он принадлежит к семейству трофических факторов, которые содержат домен эпидермального фактора роста, взаимодействуют с рецептором ErbB-4 и тесно связаны с функцией постсинаптической плотности (postsynaptic density, PSD). Ген NRG1 был признан геном чувствительности к шизофрении в разных популяциях, а понижающая регуляция nrg1 у мышей, экспрессирующих высокие уровни белка, способна противодействовать психотическим поведенческим аномалиям у этих животных. Недавно была доказана связь между NRG1 и D2R в управлении психотическим поведением у грызунов. Длительная терапия арипипразолом, оланзапином или галоперидолом может вызывать различную временную и пространственную понижающую регуляцию при разных изоформах NRG1, что подтверждает уникальные особенности функциональной селективности антипсихотических препаратов.
Недавно была доказана связь между NRG1 и D2R в управлении психотическим поведением у грызунов. Длительная терапия арипипразолом, оланзапином или галоперидолом может вызывать различную временную и пространственную понижающую регуляцию при разных изоформах NRG1, что подтверждает уникальные особенности функциональной селективности антипсихотических препаратов.
Долгосрочные эффекты применения арипипразола на дофамин-зависимую синаптическую пластичность
вверх
Хотя во многих случаях шизофрения считается нарушением, естественное течение которого трудно предсказать и которое во многом зависит от индивидуальной изменчивости, текущие данные указывают на то, что антипсихотические препараты все еще остаются основным методом лечения этого заболевания и что непрерывная терапия может предотвратить значительное число рецидивов. Более того, недостаточная приверженность или отказ от антипсихотической терапии классифицируют как явный риск рецидива с прогрессирующим ухудшением исхода. Благотворное влияние длительного лечения на патофизиологию заболевания, а также на динамические характеристики дофамина может быть в значительной степени усилено путем непрерывной терапии антипсихотиками в форме LAI (long-acting injectable, инъекционные антипсихотики длительного действия).
Благотворное влияние длительного лечения на патофизиологию заболевания, а также на динамические характеристики дофамина может быть в значительной степени усилено путем непрерывной терапии антипсихотиками в форме LAI (long-acting injectable, инъекционные антипсихотики длительного действия).
Однако доклинические исследования неоднократно сообщали о значительных, хотя часто противоречивых, различиях между транзиторным пероральным (или инъекционным) применением антипсихотиков и непрерывной инфузией с использованием соответствующих устройств (например, осмотических мининасосов, депо-препаратов). В исследованиях на людях и животных при определении действия конкретного антипсихотического препарата значительное внимание уделяется порогу занятости D2R, достигаемому этим препаратом, который коррелирует с клиническими и побочными эффектами, а также может быть полезен в дифференцировании каждого препарата и понимании его молекулярного действия.
Сравнение с другими антипсихотиками
Несмотря на огромный интерес с точки зрения потенциального клинического применения, было проведено лишь несколько исследований с целью изучения эффектов непрерывного применения арипипразола на передачу сигналов дофамина в доклинических условиях. В связи с этим сначала следует рассмотреть влияние длительного, но не непрерывного (т. е. долгосрочного ежедневного перорального приема) применения арипипразола, а затем перенести фокус на исследования, направленные на изучение эффектов долгосрочного парентерального непрерывного лечения. Сообщалось о различиях в модуляции дофаминовой нейротрансмиссии между антипсихотиками первого и нового поколения. Например, ранние исследования показали, что арипипразол может проявлять антагонистическую активность в отношении дофамина на стриатальных нейронах (т. е. незначительное повышение связывания [Н3]-спиперона и ингибирование активности ГТФазы, вызванной квинпиролом), но без увеличения экспрессии мРНК D2R, тогда как галоперидол ее значительно увеличивает. Подобным образом при долгосрочном применении арипипразола в гипофизарных клетках не наблюдались никакие эффекты на пролактин и снижение связывания спиперона, тогда как галоперидол и сульпирид существенно усиливают оба показателя.
В связи с этим сначала следует рассмотреть влияние длительного, но не непрерывного (т. е. долгосрочного ежедневного перорального приема) применения арипипразола, а затем перенести фокус на исследования, направленные на изучение эффектов долгосрочного парентерального непрерывного лечения. Сообщалось о различиях в модуляции дофаминовой нейротрансмиссии между антипсихотиками первого и нового поколения. Например, ранние исследования показали, что арипипразол может проявлять антагонистическую активность в отношении дофамина на стриатальных нейронах (т. е. незначительное повышение связывания [Н3]-спиперона и ингибирование активности ГТФазы, вызванной квинпиролом), но без увеличения экспрессии мРНК D2R, тогда как галоперидол ее значительно увеличивает. Подобным образом при долгосрочном применении арипипразола в гипофизарных клетках не наблюдались никакие эффекты на пролактин и снижение связывания спиперона, тогда как галоперидол и сульпирид существенно усиливают оба показателя.
Что касается метаболизма дофамина в переднем мозге, исследования с использованием микродиализа показали, что краткосрочная терапия арипипразолом не оказывает влияния на высвобождение дофамина ни в префронтальной коре головного мозга, ни в стриатуме, однако может значительно повышать уровни внеклеточных метаболитов дофамина и подавлять уровни внеклеточных метаболитов серотонина в обеих областях. И наоборот, долгосрочное применение арипипразола приводит к «стабилизации» дофаминергических эффектов с уменьшением концентраций дофамина и более слабым воздействием на внеклеточные метаболиты как дофамина, так и серотонина. Эти данные сильно отличаются от результатов, полученных при применении оланзапина, который существенно и постоянно увеличивает концентрацию и уровни его внеклеточных метаболитов в медиальной префронтальной коре и стриатуме. Такие эффекты выявляют присущие арипипразолу дофамин-стабилизирующие характеристики при длительном применении препарата. Более того, исследования экспрессии генов также показали, что арипипразол при длительном использовании может активировать программы экспрессии генов, существенно отличающиеся от программ, активируемых при применении других антипсихотических средств. Недавний микроматричный анализ показал, что 4-недельное применение арипипразола в клинически значимых дозах может обусловливать выборочную экспрессию таких генов, как Dnmt3a или ген катехол-O-метилтрансферазы (COMT), вовлеченных в патофизиологию психоза посредством участия в ремоделировании хроматина и в регуляции транскрипции.
И наоборот, долгосрочное применение арипипразола приводит к «стабилизации» дофаминергических эффектов с уменьшением концентраций дофамина и более слабым воздействием на внеклеточные метаболиты как дофамина, так и серотонина. Эти данные сильно отличаются от результатов, полученных при применении оланзапина, который существенно и постоянно увеличивает концентрацию и уровни его внеклеточных метаболитов в медиальной префронтальной коре и стриатуме. Такие эффекты выявляют присущие арипипразолу дофамин-стабилизирующие характеристики при длительном применении препарата. Более того, исследования экспрессии генов также показали, что арипипразол при длительном использовании может активировать программы экспрессии генов, существенно отличающиеся от программ, активируемых при применении других антипсихотических средств. Недавний микроматричный анализ показал, что 4-недельное применение арипипразола в клинически значимых дозах может обусловливать выборочную экспрессию таких генов, как Dnmt3a или ген катехол-O-метилтрансферазы (COMT), вовлеченных в патофизиологию психоза посредством участия в ремоделировании хроматина и в регуляции транскрипции. Кроме того, в одной из работ было продемонстрировано необычное воздействие арипипразола на гены синаптической пластичности, такие как Homer1a. Действительно, при краткосрочном применении только относительно низкие дозы арипипразола вызывали в стриатуме экспрессию Homer1a и его сплайсингового варианта ania-3, как и в случае галоперидола, тогда как при высоких дозах не наблюдалось таких эффектов. И наоборот, только более высокие дозы активировали кортикальную транскрипцию Homer1a.
Кроме того, в одной из работ было продемонстрировано необычное воздействие арипипразола на гены синаптической пластичности, такие как Homer1a. Действительно, при краткосрочном применении только относительно низкие дозы арипипразола вызывали в стриатуме экспрессию Homer1a и его сплайсингового варианта ania-3, как и в случае галоперидола, тогда как при высоких дозах не наблюдалось таких эффектов. И наоборот, только более высокие дозы активировали кортикальную транскрипцию Homer1a.
Длительное применение арипипразола, как и клозапина, напротив, продемонстрировало постоянную повышающую регуляцию экспрессии Homer1a в стриатуме с одновременным уменьшением его кортикальной экспрессии. Интересно, что при долгосрочном применении арипипразола в этом исследовании не наблюдалось эффектов на D2R. Эти данные еще раз подтверждают возможную балансировку синаптического высвобождения дофамина арипипразолом, который действует как на пресинаптические, так и на постсинаптические D2R и, следовательно, имеет тенденцию «стабилизировать» постсинаптическую передачу сигналов, которая нарушается при психозоподобных патологических состояниях. Дальнейшее исследование также подтвердило, что длительный прием арипипразола может ослаблять понижающую регуляцию экспрессии мозгового нейротрофического фактора (BDNF), гликогенсинтазы-киназы-3β (GSK-3beta) и β-катенина, индуцированную иммобилизирующим стрессом. С этими данными согласуются сообщения о том, что последовательное применение арипипразола на животной модели стресса с вынужденным плаванием модулировало экспрессию генов Arc и нейронного PAS-доменного белка 4 (Npsa4), коррелирующую с нейропластичностью.
Дальнейшее исследование также подтвердило, что длительный прием арипипразола может ослаблять понижающую регуляцию экспрессии мозгового нейротрофического фактора (BDNF), гликогенсинтазы-киназы-3β (GSK-3beta) и β-катенина, индуцированную иммобилизирующим стрессом. С этими данными согласуются сообщения о том, что последовательное применение арипипразола на животной модели стресса с вынужденным плаванием модулировало экспрессию генов Arc и нейронного PAS-доменного белка 4 (Npsa4), коррелирующую с нейропластичностью.
Недавно были получены сообщения о том, что непрерывная инфузия арипипразола с использованием мининасосов в течение 14 дней обеспечила поддержание уровней D2R в стриатуме крыс и не оказала влияния на локомоторные реакции при провокации метамфетамином. При этом эффекты полностью отличались от вызванных галоперидолом, который как повысил плотность D2R, так и увеличил частоту возникновения локомоторных реакций. Эти результаты подтверждают отсутствие влияния длительного приема арипипразола, даже при непрерывном применении, на сверхчувствительность к дофамину. Последняя была описана как увеличение числа D2R или усиление биологического действия D2R, которая может быть вызвана постоянным снижением активности D2R, как в случае долгосрочного лечения с использованием D2R-блокирующих агентов. Сверхчувствительность к дофамину может привести к развитию клинического состояния, известного как «сверхчувствительный» или «прорывный» психоз, представленного как антипсихотическое явление и описываемого как повторное возникновение психотических симптомов в ходе непрерывного лечения с использованием главным образом препаратов с высоким сродством к D2R. Психоз, обусловленный сверхчувствительностью, считается результатом адаптивных нейробиологических процессов дофаминовой нейротрансмиссии, возможно, увеличения числа D2R с высоким сродством (изоформ рецепторов с функциональным высоким сродством к дофамину), которые могут представлять собой подтип сверхчувствительности к дофамину, индуцированной антипсихотиками. Тем не менее, в феномен сверхчувствительности к дофамину могут быть вовлечены некоторые другие нейробиологические механизмы, включая генные мутации, черепно-мозговую травму или употребление наркотиков.
Последняя была описана как увеличение числа D2R или усиление биологического действия D2R, которая может быть вызвана постоянным снижением активности D2R, как в случае долгосрочного лечения с использованием D2R-блокирующих агентов. Сверхчувствительность к дофамину может привести к развитию клинического состояния, известного как «сверхчувствительный» или «прорывный» психоз, представленного как антипсихотическое явление и описываемого как повторное возникновение психотических симптомов в ходе непрерывного лечения с использованием главным образом препаратов с высоким сродством к D2R. Психоз, обусловленный сверхчувствительностью, считается результатом адаптивных нейробиологических процессов дофаминовой нейротрансмиссии, возможно, увеличения числа D2R с высоким сродством (изоформ рецепторов с функциональным высоким сродством к дофамину), которые могут представлять собой подтип сверхчувствительности к дофамину, индуцированной антипсихотиками. Тем не менее, в феномен сверхчувствительности к дофамину могут быть вовлечены некоторые другие нейробиологические механизмы, включая генные мутации, черепно-мозговую травму или употребление наркотиков. Было отмечено, что прорывный психоз также может быть обусловлен воздействием внешних факторов стресса у пациентов, получающих низкие дозы антипсихотиков. Следует ли связывать развитие этого состояния с рецидивом, свойственным естественному течению болезни, или с комбинацией жизненных событий и ятрогенной сверхчувствительности дофамина, является интересным вопросом для будущих исследований.
Было отмечено, что прорывный психоз также может быть обусловлен воздействием внешних факторов стресса у пациентов, получающих низкие дозы антипсихотиков. Следует ли связывать развитие этого состояния с рецидивом, свойственным естественному течению болезни, или с комбинацией жизненных событий и ятрогенной сверхчувствительности дофамина, является интересным вопросом для будущих исследований.
Несмотря на отсутствие данных, некоторые различия между кратковременным и непрерывным применением арипипразола могут быть установлены по результатам исследований поведения животных. На животных моделях самостоятельного применения кокаина и последующего ослабления и восстановления низкие дозы арипипразола, вводимые ежедневно перед каждым сеансом самостоятельного применения и восстановления, продемонстрировали уменьшение, но не предотвращение самостоятельного применения кокаина, хотя и препятствовали восстановлению поведения по его поиску. И наоборот, в случае непрерывной инфузии арипипразол не оказывает значительного влияния на самостоятельное применение кокаина или его выбор, тогда как острая инъекция быстро вызывает снижение поискового поведения, хотя только в низких промежуточных дозах. Поэтому, хотя необходимость в проведении дополнительных исследований сохраняется, эти эксперименты продемонстрировали эффект арипипразола как при остром, так и при хроническом течении шизофрении, при этом применение разных доз вызывало различные эффекты. Более того, как и в случае других антипсихотиков, транзиторное применение привело к получению дифференцированных эффектов, в отличие от непрерывной инфузии, кинетику которой еще предстоит определить. Роль длительности лечения антипсихотиками также важна для прогнозирования антипсихотического ответа и поддержания реакции. Примерно 30% пациентов не отвечают или отвечают в недостаточной степени на антипсихотическую терапию и считаются резистентными к лечению. Было высказано предположение о существовании множества факторов, являющихся источником истинной резистентности к антипсихотическим средствам, включая генетическую изменчивость реактивности рецепторов дофамина и других нейротрансмиттерных рецепторов и аберрантную морфологию головного мозга, такую как потеря серого вещества в кортикальных и подкожных областях.
Поэтому, хотя необходимость в проведении дополнительных исследований сохраняется, эти эксперименты продемонстрировали эффект арипипразола как при остром, так и при хроническом течении шизофрении, при этом применение разных доз вызывало различные эффекты. Более того, как и в случае других антипсихотиков, транзиторное применение привело к получению дифференцированных эффектов, в отличие от непрерывной инфузии, кинетику которой еще предстоит определить. Роль длительности лечения антипсихотиками также важна для прогнозирования антипсихотического ответа и поддержания реакции. Примерно 30% пациентов не отвечают или отвечают в недостаточной степени на антипсихотическую терапию и считаются резистентными к лечению. Было высказано предположение о существовании множества факторов, являющихся источником истинной резистентности к антипсихотическим средствам, включая генетическую изменчивость реактивности рецепторов дофамина и других нейротрансмиттерных рецепторов и аберрантную морфологию головного мозга, такую как потеря серого вещества в кортикальных и подкожных областях.
Предотвращение развития резистентности к лечению антипсихотиками
Одной из неудовлетворенных потребностей в контексте шизофрении, резистентной к лечению, является контроль прогрессирующей потери эффекта после успешного длительного лечения. Другими словами, после ответа на антипсихотик, применяемый в правильной дозе в течение длительного периода времени, у пациента может возникнуть рецидив с увеличением тяжести симптомов, несмотря на хорошую приверженность терапии и отсутствие модифицирующих переменных (таких как лекарственные взаимодействия или сопутствующие заболевания, которые могут поставить под угрозу лечение антипсихотиками).
Даже если механизм, лежащий в основе этого состояния, не разгадан, одна гипотеза, подкрепленная убедительными доклиническими данными, утверждает, что длительное лечение антипсихотиками с высоким сродством к D2R может постепенно индуцировать усиление связывания D2R путем увеличения числа этих рецепторов с высоким сродством (возможно также максимальное специфическое связывание D2R-Bmax), что делает D2R более чувствительными к дофамину.
Было выдвинуто интересное предположение о том, что увеличение числа D2R с высоким сродством является общим окончательным результатом множества фармакологических манипуляций (таких как применение антагонистов дофамина, агонистов дофамина, роды после кесарева сечения), даже тех, которые не имеют прямого отношения к дофаминовой системе, и что увеличение числа D2R с высоким сродством представляет собой ключевое явление для восприимчивости к развитию психозоподобного поведения на животной модели шизофрении. Если корреляция между увеличением числа D2R с высоким сродством и прогрессирующим снижением ответа на антипсихотические средства действительно подтвердится, то отсутствие увеличения количества D2R с высоким сродством после длительной терапии арипипразолом посредством непрерывной инфузии (внутрибрюшинный мининасос) по сравнению с долгосрочным лечением с использованием полных антагонистов, таких как галоперидол, может указывать на то, что смещенные лиганды способны потенциально предотвращать или задерживать (у взрослых животных, но не у молодых грызунов) предполагаемые последствия увеличения числа D2R с высоким сродством (например, развитие резистентности к антипсихотикам после периода получения надлежащего ответа).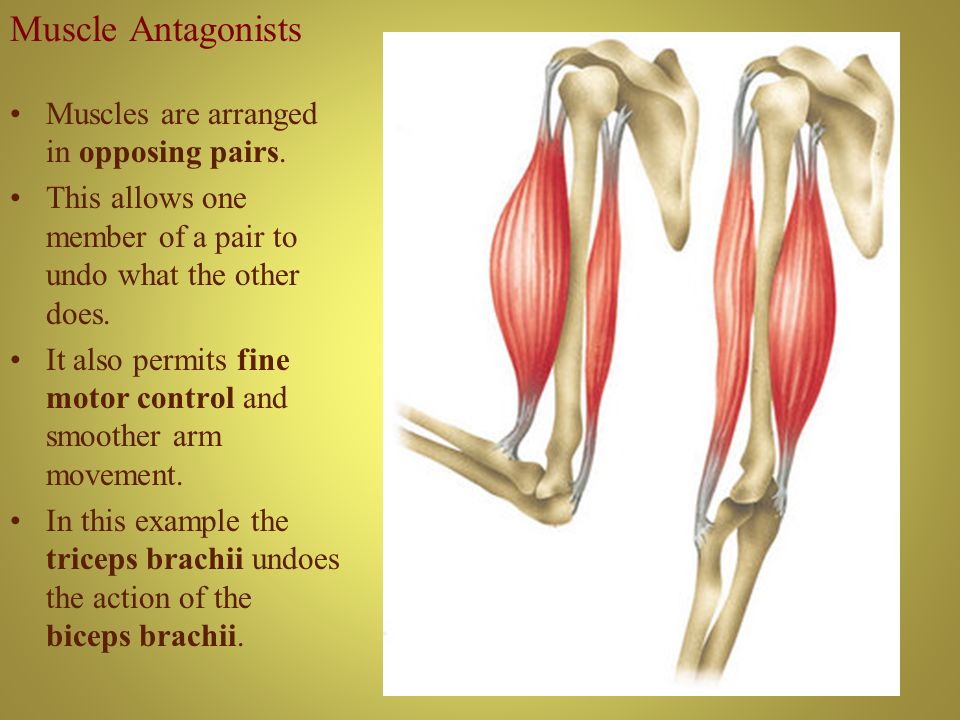 Это наблюдение следует рассматривать в совокупности с выводом о том, что многократное применение арипипразола вызывает (как и в случае других антипсихотиков, действующих на D2R) сенсибилизирующий эффект в подавлении условного рефлекса избегания (т. е. избегания раздражения лап) без нарушения реакции бегства (т. е. двигательной способности) и гиперлокомоции, индуцированной фенциклидином, – признак, который считается высокопрогностическим для антипсихотической активности.
Это наблюдение следует рассматривать в совокупности с выводом о том, что многократное применение арипипразола вызывает (как и в случае других антипсихотиков, действующих на D2R) сенсибилизирующий эффект в подавлении условного рефлекса избегания (т. е. избегания раздражения лап) без нарушения реакции бегства (т. е. двигательной способности) и гиперлокомоции, индуцированной фенциклидином, – признак, который считается высокопрогностическим для антипсихотической активности.
Заключительные комментарии
вверх
Хотя последние экспериментальные данные снижают фармакологическую значимость механизма действия антипсихотических препаратов, внедрение понятия частичного агонизма в отношении D2R стало прорывом в лечении шизофрении, поскольку это позволило под другим углом рассматривать мнение об «отсутствии эффективности без блокады D2R», которое в последние 50 лет было центральным в фармакологической терапии психоза.
Даже если рассматривать арипипразол в качестве классического частичного агониста D2R, многочисленные доказательства указывают на то, что он действует на D2R, влияя на дофаминергическую систему различными способами в зависимости от условий, в которых исследуется биологическая система. Поэтому в различных доклинических парадигмах арипипразол был описан как полный антагонист, умеренный антагонист или частичный агонист в отношении D2R, что согласуется с характеристиками смещенных лигандов. «Адаптивные» свойства арипипразола поразительно отличаются от свойств других антипсихотических препаратов, действие которых остается относительно стабильным во всех биологических условиях. Одним из основных последствий этого свойства является то, что использование арипипразола может препятствовать развитию долгосрочных нейрохимических адаптаций, связанных с дофамином, которые могут повлиять на долгосрочный ответ на антипсихотические средства. Действительно, было отмечено, что долгосрочное лечение арипипразолом предотвращает развитие сверхчувствительности к дофамину. В то время как промежуточная внутренняя активность арипипразола в отношении D2R может сама по себе служить подходящим объяснением отсутствия сверхчувствительности к дофамину, нельзя исключать, что это может зависеть от фармакологического действия арипипразола, которое варьируется от агонизма к антагонизму и поэтому колеблется между стимулированием и инактивацией рецептора.
Поэтому в различных доклинических парадигмах арипипразол был описан как полный антагонист, умеренный антагонист или частичный агонист в отношении D2R, что согласуется с характеристиками смещенных лигандов. «Адаптивные» свойства арипипразола поразительно отличаются от свойств других антипсихотических препаратов, действие которых остается относительно стабильным во всех биологических условиях. Одним из основных последствий этого свойства является то, что использование арипипразола может препятствовать развитию долгосрочных нейрохимических адаптаций, связанных с дофамином, которые могут повлиять на долгосрочный ответ на антипсихотические средства. Действительно, было отмечено, что долгосрочное лечение арипипразолом предотвращает развитие сверхчувствительности к дофамину. В то время как промежуточная внутренняя активность арипипразола в отношении D2R может сама по себе служить подходящим объяснением отсутствия сверхчувствительности к дофамину, нельзя исключать, что это может зависеть от фармакологического действия арипипразола, которое варьируется от агонизма к антагонизму и поэтому колеблется между стимулированием и инактивацией рецептора.
Другим значимым последствием «адаптивного» свойства арипипразола является то, что фармакологическое действие соединения может во многом зависеть, помимо других механизмов, от уровней эндогенного дофамина и статуса сигнализации.
В заключение стоит отметить, что включение арипипразола в схему терапии при шизофрении и других психических заболеваниях проложило путь для нового поколения соединений, которые не только блокируют передачу дофамина через D2R, но также могут быть представлены в качестве динамических модуляторов D2R за пределами дофаминового антагонизма.
Список литературы
1. Bartolomeis A. et al. Update on the Mechanism of Action of Aripiprazole: Translational Insights into Antipsychotic Strategies Beyond Dopamine Receptor Antagonism. CNS Drugs. 2015; 29: 773-799.
2. Gründer G. et al. Brain and Plasma Pharmacokinetics of Aripiprazole in Patients With Schizophrenia: An [18F] Fallypride PET Study. Am J Psychiatry. 2008; 165: 988-995.
Am J Psychiatry. 2008; 165: 988-995.
3. Burris K.D. et al. Aripiprazole, a Novel Antipsychotic, Is a High-Affinity Partial Agonist at Human Dopamine D2 Receptors. J Pharmacol Exp Ther. 2002; 302 (1): 381-9.
4. Kane J.M. et al. Symptomatic remission in schizophrenia patients treated with aripiprazole or haloperidol for up to 52 weeks. Schizophr Res. 2007; 95 (1-3): 143-50.
NP-PSYC-PUB-052017-006
фармакодинамика, агонизм и антагонизм, специфичность препаратов. Теории взаимодействия лекарственных препаратов с рецепторами
Агонисты рецепторов — лекарственные вещества, стимулирующие рецепторы, сходны с природными медиаторами или гормонами. Их ценность при патологических состояниях состоит в том, что они обладают большей устойчивостью, чем истинные медиаторы, по отношению к разрушающим веществам, поэтому их действие продолжительнее действия природных веществ, эффекты которых они имитируют.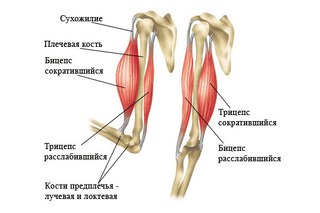
В начале настоящего столетия было высказано предположение о том, что некоторые лекарства вызывают эффекты в результате связывания со специализированными рецепторами на клетках. Тот факт, что кураре устраняет сокращение мышцы, вызванное никотином, и не предотвращает сокращение, вызванное ее электростимуляцией, позволил Langley (1905) сделать вывод о том, что то и другое вещество действует в результате образования комплекса с определенным компонентом на мышечной клетке, но не с сократительным веществом. Его назвали рецепторным веществом, или рецептором . В последующие годы понятие «рецептор» использовали как основу для концепции, позволяющей не только обсуждать механизмы действия известных лекарств, но и осуществлять поиск новых. Количественное определение рецепторов и изучение их распределения стали возможными после того, как было установлено, что яд змеи α-токсин (в который можно внести радиоактивную метку) избирательно связывается с ацетилхолиновыми рецепторами на синапсах в скелетной мускулатуре. С помощью электронной микрофотографии в этой ткани были выявлены рецепторные молекулы. Стало очевидным, что классическая концепция, в соответствии с которой взаимосвязь вещества и рецептора рассматривали как «ключ и замок», слишком ограниченна. Метод с использованием радиолигандов позволяет определять количество и изучать связывающую способность рецепторов как на целых клетках, так и в препаратах, приготовленных из их оболочек.
С помощью электронной микрофотографии в этой ткани были выявлены рецепторные молекулы. Стало очевидным, что классическая концепция, в соответствии с которой взаимосвязь вещества и рецептора рассматривали как «ключ и замок», слишком ограниченна. Метод с использованием радиолигандов позволяет определять количество и изучать связывающую способность рецепторов как на целых клетках, так и в препаратах, приготовленных из их оболочек.
Было установлено, что количество рецепторов не постоянно , а изменяется при разных обстоятельствах. Оно уменьшается при длительном воздействии на ткань антагониста, что может привести к развитию тахифилаксни, т.е. к утрате эффективности при повторном частом применении препарата, например бронхорасширяющего действия симпатомиметиков при бронхиальной астме. Длительное воздействие агониста сопровождается образованием новых рецепторов. В связи с этим быстрая отмена бета-адреноблокаторов у больных, страдающих стенокардией или аритмией, может сопровождаться ухудшением состояния, так как катехоламины, циркулирующие в крови, оказывают более сильное действие в связи с увеличением количества бета-адренорецепторов.
Большинство рецепторов представляет собой белковые молекулы. При связывании с рецептором агониста изменяется конформация белковой молекулы, что сопровождается изменением внутриклеточных процессов, определяющих реакцию на лекарственный препарат. Например, активация бета-адренорецепторов катехоламином (первичный мессенжер) повышает активность аденилатциклазы, ускоряющей образование цАМФ (вторичный мессенжер), регулирующего активность нескольких ферментативных систем, активирующих клетку. Влияние лекарственного вещества на рецептор может опосредоваться также воздействием на функцию мембранных ионных каналов, тесно связанных с рецептором (например, с никотиночувствительным ацетилхолиновым рецептором), или изменением уровня внутриклеточного кальция (например, реализация некоторых эффектов через мускариночувствительные рецепторы).
Агонисты рецепторов
Агонисты рецепторов — лекарственные вещества, стимулирующие рецепторы, сходны с природными медиаторами или гормонами. Их ценность при патологических состояниях состоит в том, что они обладают большей устойчивостью, чем истинные медиаторы, по отношению к разрушающим веществам, поэтому их действие продолжительнее действия природных веществ, эффекты которых они имитируют. Например, бронхорасширяющее действие сальбутамола продолжительнее, чем адреналина.
Их ценность при патологических состояниях состоит в том, что они обладают большей устойчивостью, чем истинные медиаторы, по отношению к разрушающим веществам, поэтому их действие продолжительнее действия природных веществ, эффекты которых они имитируют. Например, бронхорасширяющее действие сальбутамола продолжительнее, чем адреналина.
Антагонисты рецепторов
Антагонисты (блокаторы) рецепторов близки к природным агонистам, поэтому рецептор «узнает» их, но, занимая рецептор, антагонисты не активируют его, при этом и естественный агонист не может активировать его. Лекарственные средства, занимающие и не активирующие рецептор, называются чистыми антагонистами .
Частичные агонисты. Некоторые лекарства, блокирующие рецептор, способны частично и стимулировать его, т. е. обладают свойствами как антагониста, так и агониста. Их эффекты зависят от обстоятельств, например налорфин в средних дозах выступает в роли антагониста опиоидов по отношению к их угнетающему действию на дыхательный центр, но в больших дозах может усиливать угнетение дыхания. В связи с этим с появлением налоксона, чистого антагониста, налорфин утратил свое клиническое значение. Такие вещества, как пентазоцин, называют средствами со свойствами частичных агонистов. Пиндолол и окспренолол относятся к бета-адреноблокаторам, обладающим этими свойствами. Их нередко называют бета-блокаторами с внутренней симпатомиметической активностью в отличие от пропранолола (анаприлин), не обладающего свойствами агониста и поэтому представляющего собой чистый антагонист. Разница между ними по клиническим признакам довольно существенна, так как у больных в состоянии покоя пропранолол (анаприлин) заметнее урежает пульс в отличие от пиндолола, несмотря на то что оба препарата защищают организм от воздействия изменяющейся концентрации катехоламинов в крови, так как блокируют бета-рецептор. В связи с этим при физических нагрузках и тот и другой препарат в равной мере нивелирует рефлекторную тахикардию. Возможно, что при определенных условиях такие различия в действии могут иметь терапевтическое значение.
В связи с этим с появлением налоксона, чистого антагониста, налорфин утратил свое клиническое значение. Такие вещества, как пентазоцин, называют средствами со свойствами частичных агонистов. Пиндолол и окспренолол относятся к бета-адреноблокаторам, обладающим этими свойствами. Их нередко называют бета-блокаторами с внутренней симпатомиметической активностью в отличие от пропранолола (анаприлин), не обладающего свойствами агониста и поэтому представляющего собой чистый антагонист. Разница между ними по клиническим признакам довольно существенна, так как у больных в состоянии покоя пропранолол (анаприлин) заметнее урежает пульс в отличие от пиндолола, несмотря на то что оба препарата защищают организм от воздействия изменяющейся концентрации катехоламинов в крови, так как блокируют бета-рецептор. В связи с этим при физических нагрузках и тот и другой препарат в равной мере нивелирует рефлекторную тахикардию. Возможно, что при определенных условиях такие различия в действии могут иметь терапевтическое значение.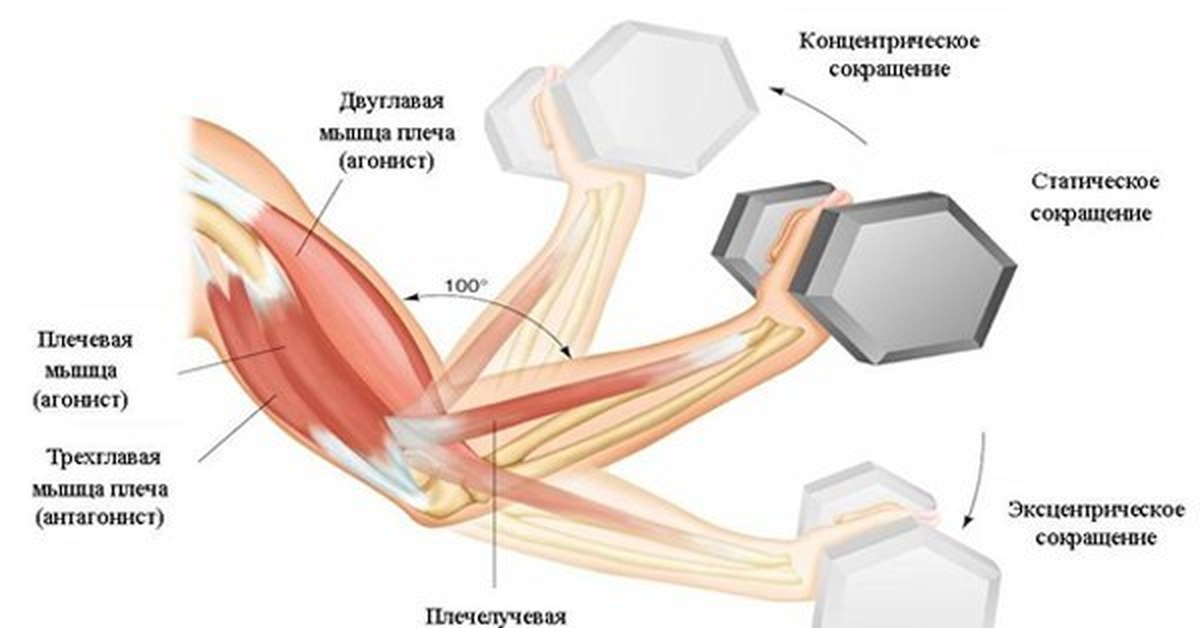
Связь с рецептором
Если связывание с рецептором слабое (водородные, вандерваальсовы, или электростатические, связи), то оно обратимо, если же прочное (ковалентное), то необратимо.
Антагонист, обратимо связывающийся с рецептором , может быть вытеснен из этой связи по закону действующих масс, согласно которому скорость химической реакции пропорциональна концентрации реагирующих веществ. При увеличении концентрации агониста в достаточной степени (конкуренция) реакция рецептора восстанавливается. Это явление нередко наблюдают в клинической практике. Если у больного, принимающего бета-адреноблокаторы, при физической нагрузке учащается пульс по сравнению с его частотой в покое, это свидетельствует о способности его симпатической нервной системы высвобождать то количество норадреналина (агонист), которое устраняет блокирующее действие использованной дозы препарата. Увеличение дозы бета-адреноблокатора может ограничить или даже полностью нивелировать тахикардию, вызванную физической нагрузкой, что свидетельствует о большей выраженности блокады, которая усилилась из-за большого количества препарата, способного конкурировать с эндогенным медиатором. Поскольку агонист и антагонист конкурируют за связь с рецептором по закону действующих масс, эти взаимоотношения лекарств получили название конкурентного антагонизма (например, использование конкурентных антагонистов при передозировке бета-адреноблокаторов). При графическом изображении зависимости эффекта от логарифма дозы и использованием данных изучения реакции на агонист, опосредуемой через рецептор на изолированных тканях, или по функциональной реакции организма, получают S-образную (сигмоидная) кривую, центральная часть которой образует прямую линию. Если измерения проводятся в присутствии антагониста и кривая, параллельная первой, смещается вправо, это свидетельствует о конкурентном взаимодействии агониста и антагониста — конкурентный антагонизм .
Поскольку агонист и антагонист конкурируют за связь с рецептором по закону действующих масс, эти взаимоотношения лекарств получили название конкурентного антагонизма (например, использование конкурентных антагонистов при передозировке бета-адреноблокаторов). При графическом изображении зависимости эффекта от логарифма дозы и использованием данных изучения реакции на агонист, опосредуемой через рецептор на изолированных тканях, или по функциональной реакции организма, получают S-образную (сигмоидная) кривую, центральная часть которой образует прямую линию. Если измерения проводятся в присутствии антагониста и кривая, параллельная первой, смещается вправо, это свидетельствует о конкурентном взаимодействии агониста и антагониста — конкурентный антагонизм .
Феноксибензамин — препарат, необратимо связывающийся альфа-адренорецепторами. Поскольку он не может быть вытеснен из рецептора, увеличение концентрации агониста не может полностью восстановить реакцию на стимуляцию рецептора. Антагонизм этого типа называют необратимым. Кривые, графически изображающие логарифм доза — эффект для агониста в отсутствие и в присутствии неконкурирующего антагониста, не параллельны. Таким образом, действуют некоторые токсины, например альфа-бунгаротоксин, входящий в состав яда некоторых змей, необратимо связывает рецептор ацетилхолина и поэтому применяется в фармакологических исследованиях. Нормализация реакции после необратимого связывания происходит только после элиминации лекарственного вещества из организма и синтеза новых рецепторов, поэтому действие подобных веществ продолжается и после прекращения их введения.
Антагонизм этого типа называют необратимым. Кривые, графически изображающие логарифм доза — эффект для агониста в отсутствие и в присутствии неконкурирующего антагониста, не параллельны. Таким образом, действуют некоторые токсины, например альфа-бунгаротоксин, входящий в состав яда некоторых змей, необратимо связывает рецептор ацетилхолина и поэтому применяется в фармакологических исследованиях. Нормализация реакции после необратимого связывания происходит только после элиминации лекарственного вещества из организма и синтеза новых рецепторов, поэтому действие подобных веществ продолжается и после прекращения их введения.
Механизм действия лекарств, основанный на регуляции рецепторов
Реакцию клеток обеспечивают рецепторы. В структуру и химические свойства каждого химического регулятора встроена определенная биологическая информация. Для того чтобы она могла быть воспринята клеткой, она должна быть расшифрована, подобно тому как радиоприемник расшифровывает именно те радиоволны, на которые он настроен. Рецептор в определенной степени напоминает активный участок фермента, т.е. он представляет собой макромолекулярный участок, по своей конфигурации и распределению ионных зарядов комплементарный соответствующему гормону. Однако в то время как в субстрате при взаимодействии с ферментом происходят химические изменения, а фермент не изменяется, гормон при взаимодействии с рецептором также не изменяется, но их взаимодействие приводит к изменению рецептора. После изменений в строении и распределении зарядов в рецепторе начинается направленное изменение клеточной активности.
Рецептор в определенной степени напоминает активный участок фермента, т.е. он представляет собой макромолекулярный участок, по своей конфигурации и распределению ионных зарядов комплементарный соответствующему гормону. Однако в то время как в субстрате при взаимодействии с ферментом происходят химические изменения, а фермент не изменяется, гормон при взаимодействии с рецептором также не изменяется, но их взаимодействие приводит к изменению рецептора. После изменений в строении и распределении зарядов в рецепторе начинается направленное изменение клеточной активности.
Подобно ферментам, рецепторы служат обычным местом приложения действия лекарственных веществ. В физиологических условиях на ферменты и рецепторы направляются селективные химические воздействия, и, если в препарате оказывается достаточно понятная клеткам информация, он сможет «обмануть» механизм регуляции организма. Подобно тому, как ингибиторы ферментов, например аллопуринол, часто очень сходны по химическому составу с обычным субстратом, антагонисты рецепторов сходны с природными гормонами.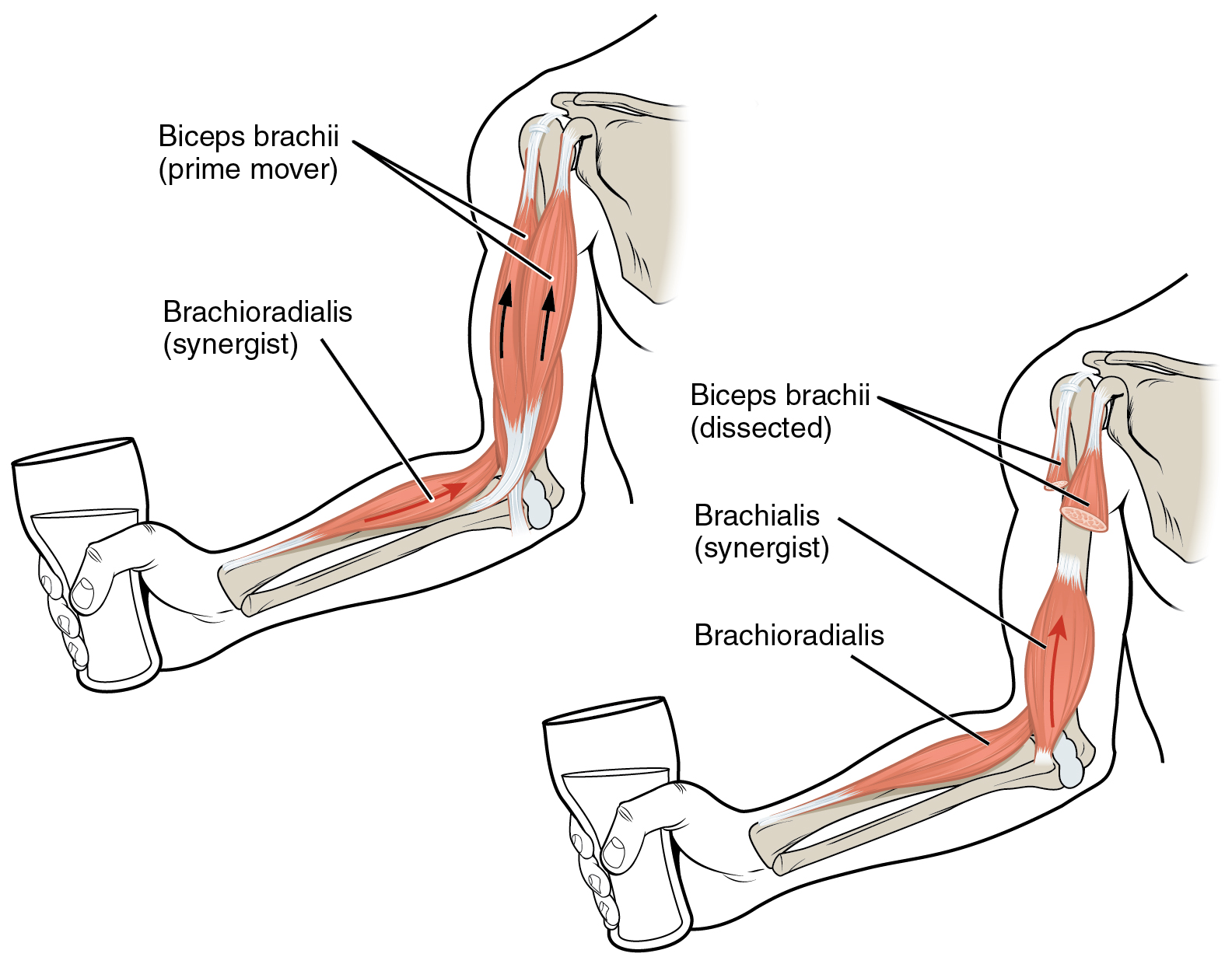 Понимание физиологической функции специфической системы гормон — рецептор может быть использовано для того, чтобы предположить, какими свойствами должно обладать новое вещество, вмешивающееся в механизмы регуляции (антагонист). Известны многочисленные примеры спекуляций такого рода, но известен и случай, приведший к созданию пропранолола (индерал, анаприлин), эффективного при заболевании сердца и выраженной гипертензии.
Понимание физиологической функции специфической системы гормон — рецептор может быть использовано для того, чтобы предположить, какими свойствами должно обладать новое вещество, вмешивающееся в механизмы регуляции (антагонист). Известны многочисленные примеры спекуляций такого рода, но известен и случай, приведший к созданию пропранолола (индерал, анаприлин), эффективного при заболевании сердца и выраженной гипертензии.
Человек может жить несколько месяцев без пищи и несколько дней без воды, но не способен выдержать даже несколько минут без воздуха и кислорода. После прекращения подачи кислорода прежде всего страдает сердце. Он поступает в сердечную мышцу по артериям, через которые кровь проходит главным образом во время короткой паузы между сокращениями. Доставка кислорода настолько важна, что у сердца есть собственное специальное кровоснабжение — коронарные артерии. Без кислорода сердечная мышца перестает сокращаться и погибает. Коронарные артерии представляют собой собственную систему жизнеобеспечения сердца. На повышение активности организма, будь то физическая нагрузка или возбуждение, сердце реагирует повышением частоты и интенсивности сокращений, что обусловлено выделением норадреналина специальными (симпатические) нервными окончаниями в волокнах сердечной мышцы. Для того чтобы выполнить эту дополнительную работу, сердцу требуется больше кислорода, поэтому коронарные артерии должны доставлять кровь быстрее. В норме артерии осуществляют это, подобно водопроводному крану, расширяя свой просвет. Однако при заболевании на внутренней оболочке артерий появляются утолщения, что обусловливает сужение их отверстий до такой степени, что ток крови не может усилиться и обеспечить потребности организма в кислороде (как при образовании налета в водопроводной трубе: сколько ни открывай кран, поток воды не усилится). Первый признак неблагополучия человек ощущает при неспособности коронарных артерий обеспечить потребность сердца в крови и кислороде, например во время интенсивной физической нагрузки. В критический момент, когда потребность в кислороде превышает обеспечение им, возникает боль, которая может быть очень сильной, — так начинается приступ стенокардии.
На повышение активности организма, будь то физическая нагрузка или возбуждение, сердце реагирует повышением частоты и интенсивности сокращений, что обусловлено выделением норадреналина специальными (симпатические) нервными окончаниями в волокнах сердечной мышцы. Для того чтобы выполнить эту дополнительную работу, сердцу требуется больше кислорода, поэтому коронарные артерии должны доставлять кровь быстрее. В норме артерии осуществляют это, подобно водопроводному крану, расширяя свой просвет. Однако при заболевании на внутренней оболочке артерий появляются утолщения, что обусловливает сужение их отверстий до такой степени, что ток крови не может усилиться и обеспечить потребности организма в кислороде (как при образовании налета в водопроводной трубе: сколько ни открывай кран, поток воды не усилится). Первый признак неблагополучия человек ощущает при неспособности коронарных артерий обеспечить потребность сердца в крови и кислороде, например во время интенсивной физической нагрузки. В критический момент, когда потребность в кислороде превышает обеспечение им, возникает боль, которая может быть очень сильной, — так начинается приступ стенокардии. Состояние мышцы сердца может нормализоваться при прекращении дополнительной нагрузки из-за возникшей боли. Таким образом, работа сердца уменьшается до уровня, который коронарные артерии в состоянии поддерживать. Активность больного в этом случае оказывается ограниченной. Однако в каком-нибудь участке мышцы сердца могут произойти необратимые изменения. Таким образом, развивается инфаркт миокарда. После довольно распространенного инфаркта неповрежденные участки сердечной мышцы все еще могут поддерживать необходимый уровень ее насосной функции при условии, что в нервных окончаниях выделяется достаточное количество норадреналина. Печальный парадокс инфаркта сердца состоит в том, что стимуляция норадреналином, необходимая для поддержания адекватного сокращения, увеличивает и вероятность нарушения стимуляции сердечной мышцы на границе неповрежденного и поврежденного участков. Такая аномальная стимуляция может нарушить координированное сокращение сердца: его стенка начинает сокращаться судорожно и несинхронно и внезапно теряет способность быть эффективным насосом.
Состояние мышцы сердца может нормализоваться при прекращении дополнительной нагрузки из-за возникшей боли. Таким образом, работа сердца уменьшается до уровня, который коронарные артерии в состоянии поддерживать. Активность больного в этом случае оказывается ограниченной. Однако в каком-нибудь участке мышцы сердца могут произойти необратимые изменения. Таким образом, развивается инфаркт миокарда. После довольно распространенного инфаркта неповрежденные участки сердечной мышцы все еще могут поддерживать необходимый уровень ее насосной функции при условии, что в нервных окончаниях выделяется достаточное количество норадреналина. Печальный парадокс инфаркта сердца состоит в том, что стимуляция норадреналином, необходимая для поддержания адекватного сокращения, увеличивает и вероятность нарушения стимуляции сердечной мышцы на границе неповрежденного и поврежденного участков. Такая аномальная стимуляция может нарушить координированное сокращение сердца: его стенка начинает сокращаться судорожно и несинхронно и внезапно теряет способность быть эффективным насосом. Это состояние называется фибрилляцией сердца, которое обычно приводит к внезапной смерти, однако оказание срочной помощи (электрический шок, вызываемый дефибриллятором) способствует нормализации ритма.
Это состояние называется фибрилляцией сердца, которое обычно приводит к внезапной смерти, однако оказание срочной помощи (электрический шок, вызываемый дефибриллятором) способствует нормализации ритма.
Традиционно больных стенокардией лечат нитратами, а инфарктом миокарда — покоем и анальгетиками. Нитраты вызывают ощущение тепла, покраснение лица. Считают, что аналогичное расширение кровеносных сосудов происходит и в сердечной мышце, что может обеспечить поступление в нее большего количества крови. Широкий поиск лекарственных препаратов, обладающих наиболее выраженными свойствами расширять коронарные сосуды, более избирательно и длительно действующих, оказался довольно успешным. Новейшие препараты действительно увеличивают коронарный кровоток, но часто оказываются не в состоянии предотвратить или облегчить приступ стенокардии! В этом, по-видимому, нет ничего удивительного: пораженные артерии не могут расширяться так же, как интактные. Препарат действительно может увеличить снабжение кровью сердечной мышцы, вызывая изменения нервных рефлексов. Такие рефлекторные воздействия могут привести к увеличению потребности миокарда в кислороде. Если же невозможно с помощью лекарственного препарата эффективно увеличить подачу кислорода, почему бы не попытаться уменьшить потребность в нем сердечной мышцы? Именно это происходит, когда больной при приступах стенокардии дает себе отдых или же больной с инфарктом соблюдает постельный режим. Проблема состоит в том, что стимуляция сердца норадреналином, в основном определяющая его потребность в кислороде, лишь частично контролируется физической нагрузкой; возбуждение, страх, боль или даже физический дискомфорт также стимулируют его функцию. Просто отдыха оказывается недостаточно. Возникает идея поиска таких препаратов, которые могли бы предотвратить воздействие норадреналина и таким образом контролировать потребность сердечной мышцы в кислороде.
Такие рефлекторные воздействия могут привести к увеличению потребности миокарда в кислороде. Если же невозможно с помощью лекарственного препарата эффективно увеличить подачу кислорода, почему бы не попытаться уменьшить потребность в нем сердечной мышцы? Именно это происходит, когда больной при приступах стенокардии дает себе отдых или же больной с инфарктом соблюдает постельный режим. Проблема состоит в том, что стимуляция сердца норадреналином, в основном определяющая его потребность в кислороде, лишь частично контролируется физической нагрузкой; возбуждение, страх, боль или даже физический дискомфорт также стимулируют его функцию. Просто отдыха оказывается недостаточно. Возникает идея поиска таких препаратов, которые могли бы предотвратить воздействие норадреналина и таким образом контролировать потребность сердечной мышцы в кислороде.
Норадреналиновые рецепторы представляют собой особые участки клеток сердечной мышцы, которые первыми «узнают» норадреналин и соединяются с ним, а затем изменяют клеточные ферменты, «заставляющие» сердце сокращаться быстрее и сильнее. Было выявлено, что пропранолол (анаприлин) «узнается» норадреналиновыми рецепторами клетки сердца и связывается с ними; при этом он не только не действует на механизмы, вызывающие повышение ферментативной активности в сердце, но и, связываясь с рецептором, предотвращает подобное действие норадреналина. Уже этого свойства пропранолола могло бы быть достаточно, чтобы сделать его полезным препаратом, но было выявлено еще одно исключительно важное его свойство. Норадреналиновые рецепторы в кровеносных сосудах, по-видимому, отличаются от рецепторов сердечной мышцы. Первые в настоящее время относят преимущественно к альфа-адренорецепторам, тогда как пропранолол оказался избирательным антагонистом бета-адренорецепторов сердечной мышцы. Это означает, что он эффективен при изменениях, возникающих в сердце при физической или эмоциональной нагрузке, но практически не действует на нервную регуляцию по отношению к кровеносным сосудам. При физической нагрузке нервные окончания в них в результате выделения норадреналина обусловливают перераспределение крови, которая из кожи и внутренних органов начинает поступать к мышцам, увеличивая их кровоснабжение.
Было выявлено, что пропранолол (анаприлин) «узнается» норадреналиновыми рецепторами клетки сердца и связывается с ними; при этом он не только не действует на механизмы, вызывающие повышение ферментативной активности в сердце, но и, связываясь с рецептором, предотвращает подобное действие норадреналина. Уже этого свойства пропранолола могло бы быть достаточно, чтобы сделать его полезным препаратом, но было выявлено еще одно исключительно важное его свойство. Норадреналиновые рецепторы в кровеносных сосудах, по-видимому, отличаются от рецепторов сердечной мышцы. Первые в настоящее время относят преимущественно к альфа-адренорецепторам, тогда как пропранолол оказался избирательным антагонистом бета-адренорецепторов сердечной мышцы. Это означает, что он эффективен при изменениях, возникающих в сердце при физической или эмоциональной нагрузке, но практически не действует на нервную регуляцию по отношению к кровеносным сосудам. При физической нагрузке нервные окончания в них в результате выделения норадреналина обусловливают перераспределение крови, которая из кожи и внутренних органов начинает поступать к мышцам, увеличивая их кровоснабжение.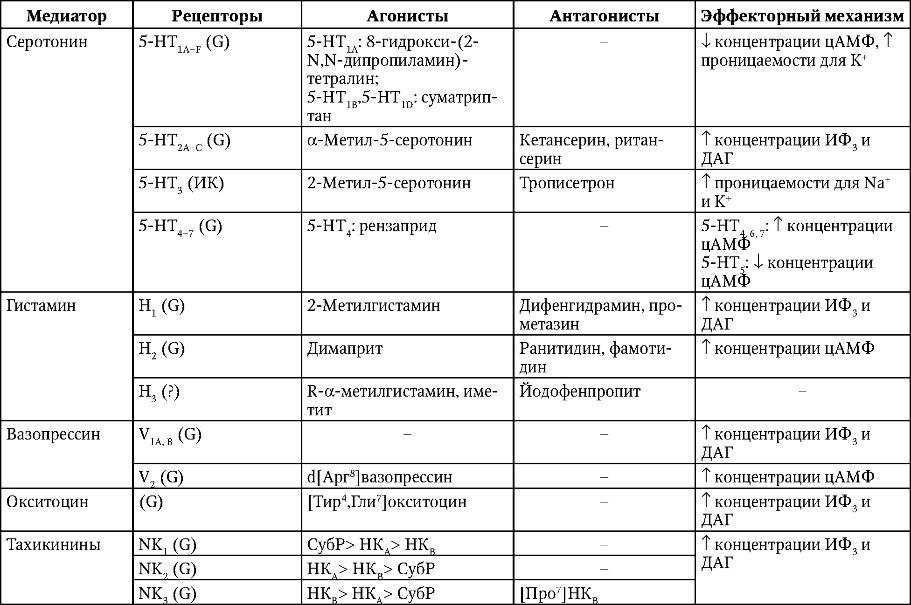 Пропранолол не влияет на это действие норадреналина, поскольку бета-адренорецепторы практически не участвуют в этом процессе.
Пропранолол не влияет на это действие норадреналина, поскольку бета-адренорецепторы практически не участвуют в этом процессе.
У больных с коронарной недостаточностью, получающих пропранолол, большая физическая нагрузка может не сопровождаться болью. Есть также данные о том, что длительная блокада бета-адренорецепторов увеличивает продолжительность жизни. Приятным сюрпризом при клинических испытаниях оказалась эффективность пропранолола при выраженной гипертензии. Если его действие также связано со способностью снижать работу сердца и сердечный выброс во время физической нагрузки (что кажется вероятным), это прольет свет на возможное происхождение этого широко распространенного заболевания.
Таким образом, речь идет о препарате, который не только приносит облегчение больным, но и дает очень много оснований для понимания роли норадреналина и родственных гормонов как в норме, так и при патологии. В настоящее время это один из важнейших аспектов изучения лекарственных средств. Они не просто приносят облегчение больному, а служат важными инструментами медицинских исследований, помогая понимать природу заболевания.
Они не просто приносят облегчение больному, а служат важными инструментами медицинских исследований, помогая понимать природу заболевания.
Другим примером действенного использования новых препаратов служит гистамин. Блокаторы бета-адренорецепторов были открыты после того, как обнаружилось, что существующие антагонисты (блокаторы альфа-адренорецепторов) не способны предупреждать реакцию сердечной мышцы на адреналин. Новые блокаторы гистамина были открыты после выявления неспособности старых противогистаминных средств предупреждать реакцию на него желез слизистой оболочки желудка. Она секретирует соляную кислоту, которая, вопреки распространенному мнению, по-видимому, играет значительно меньшую роль в пищеварении, чем в стерилизации верхнего отдела кишечника. Например, уровень заболеваемости туберкулезом выше у лиц, у которых соляная кислота не секретируется. Каждый раз во время еды начинается секреция кислоты в желудке. У некоторых лиц секретируется слишком большое ее количество в связи с тем, что раздражитель чрезвычайно силен или, возможно, из-за дефекта в механизме, прекращающего секрецию при окончании процесса пищеварения. В любом случае избыточная секреция соляной кислоты сопряжена с риском развития язвы желудка или двенадцатиперстной кишки. Эти язвы (пептические) могут причинить массу беспокойств из-за боли и недостаточности пищеварения или же привести к серьезным, даже летальным осложнениям: сильному кровотечению или прободению стенки желудка, при котором его содержимое попадает в брюшную полость, в результате чего развивается перитонит. Эти железы слизистой оболочки желудка можно устранить хирургическим путем, блокировав таким образом секрецию кислоты; можно также перерезать нервы, что прекращает стимуляцию секреции. Однако после этих операций нередко наступает летальный исход (более 1:200).
В любом случае избыточная секреция соляной кислоты сопряжена с риском развития язвы желудка или двенадцатиперстной кишки. Эти язвы (пептические) могут причинить массу беспокойств из-за боли и недостаточности пищеварения или же привести к серьезным, даже летальным осложнениям: сильному кровотечению или прободению стенки желудка, при котором его содержимое попадает в брюшную полость, в результате чего развивается перитонит. Эти железы слизистой оболочки желудка можно устранить хирургическим путем, блокировав таким образом секрецию кислоты; можно также перерезать нервы, что прекращает стимуляцию секреции. Однако после этих операций нередко наступает летальный исход (более 1:200).
Окончания нервов, которые перерезает хирург, секретируют ацетилхолин. Следовательно, и атропин, конкурентный антагонист ацетилхолина, должен вызывать тот же эффект. В течение многих лет он и родственные ему препараты применяли при пептических язвах желудка, но результаты были разочаровывающими. Дозы атропина, необходимые для уменьшения секреции соляной кислоты, блокируют и другие рецепторы ацетилхолина, вызывая нечеткость зрения, сухость слизистых оболочек в полости рта и затруднения при мочеиспускании. Он блокирует также рецепторы нервов, идущих к мышцам стенки желудка, так что эвакуация из него замедляется. Все это нивелирует положительное воздействие атропина в отношении снижения кислотности желудочного сока. Его действие недостаточно избирательно.
Он блокирует также рецепторы нервов, идущих к мышцам стенки желудка, так что эвакуация из него замедляется. Все это нивелирует положительное воздействие атропина в отношении снижения кислотности желудочного сока. Его действие недостаточно избирательно.
Помимо ацетилхолина, в желудке обнаружены два других вещества, представляющих собой мощные стимуляторы его секреции: гистамин и гастрин, Гастрин (полипептид), высвобождаемый из пищи в другом отделе кишечника, служит основным гормоном, контролирующим секрецию в желудке. Он достигает своих рецепторов на клетках слизистой оболочки желудка, поступая к ним из крови, стимулирует секрецию как переваривающих ферментов, так и соляной кислоты. Гистамин происходит из одной аминокислоты (гистидин) и сконцентрирован в области секретирующих кислоту клеток. Он лишь стимулирует секрецию соляной кислоты. Некоторые исследователи считают, что эта стимуляция опосредована локальным высвобождением гистамина из его депо.
Подобно норадреналину, гистамин действует на рецепторы двух типов: h2 и Н2. Противогистаминные средства, используемые при сенной лихорадке, блокируют Н1-рецепторы. Несколько лет назад были найдены антагонисты, блокирующие Н2-рецепторы. Один из них [циметидин (тагамет) ] в настоящее время применяется в клинике. Несмотря на то, что он представляет собой конкурентный антагонист гистамина, он подавляет также действие гастрина. Это приятная новость для больных с пептической язвой желудка, поскольку секрецию кислоты в нем в настоящее время можно избирательно уменьшить. Большее число больных могут быстрее излечиться, чем при использовании препаратов, блокирующих эффект ацетилхолина. У них есть шанс избежать серьезных осложнений, сопровождающих хирургическое вмешательство. В 1985 г., почти через 10 лет интенсивного клинического использования циметидина, у него не было обнаружено неожиданных побочных эффектов. Медицинские работники получили при этом новый инструмент для изучения функции гистамина. Учитывая, что он играет защитную роль при воспалении и восстановлении поврежденных тканей, можно полагать, что и секреция
Противогистаминные средства, используемые при сенной лихорадке, блокируют Н1-рецепторы. Несколько лет назад были найдены антагонисты, блокирующие Н2-рецепторы. Один из них [циметидин (тагамет) ] в настоящее время применяется в клинике. Несмотря на то, что он представляет собой конкурентный антагонист гистамина, он подавляет также действие гастрина. Это приятная новость для больных с пептической язвой желудка, поскольку секрецию кислоты в нем в настоящее время можно избирательно уменьшить. Большее число больных могут быстрее излечиться, чем при использовании препаратов, блокирующих эффект ацетилхолина. У них есть шанс избежать серьезных осложнений, сопровождающих хирургическое вмешательство. В 1985 г., почти через 10 лет интенсивного клинического использования циметидина, у него не было обнаружено неожиданных побочных эффектов. Медицинские работники получили при этом новый инструмент для изучения функции гистамина. Учитывая, что он играет защитную роль при воспалении и восстановлении поврежденных тканей, можно полагать, что и секреция
соляной кислоты в желудке может рассматриваться как часть защитной системы. И все же присутствие гистамина в мозге приводит в недоумение. Он синтезируется в мозге, но функция его неясна. Однако в настоящее время, когда стало возможно различать гистаминовые рецепторы, применяя препараты, подобные циметидину, может наступить прогресс в создании новых лекарственных средств.
И все же присутствие гистамина в мозге приводит в недоумение. Он синтезируется в мозге, но функция его неясна. Однако в настоящее время, когда стало возможно различать гистаминовые рецепторы, применяя препараты, подобные циметидину, может наступить прогресс в создании новых лекарственных средств.
Все больше и больше начинают понимать, каким образом лекарственные вещества «используют» механизмы контроля организма (рецепторы, гормоны, места связывания и др.) для обеспечения избирательности действия. Это понимание обеспечивает надежду в будущем получить еще большее число совершенно новых препаратов, что позволит увеличить число больных, получивших пользу от современной медицины.
Однако никакое искусство при получении более избирательно действующих и эффективных препаратов не уведет исследователя от проблемы их токсичности. Это бесспорная истина. Взаимодействие препарата с его мишенью определяется его соответствием активному месту на клеточной оболочке и вероятностью того, что хаотичное движение молекул способствует контакту молекулы препарата с активным участком. Возможность этого контакта определяется главным образом количеством молекул. В тех случаях, когда молекулы препарата отличаются высоким сродством к активным участкам на клеточной оболочке, даже небольшое их число может обеспечить взаимодействие. Однако по мере увеличения концентрации препарата возможно эффективное его взаимодействие с активными участками, имеющими меньшее сродство к препарату. Это может обусловить его дополнительные эффекты, в том числе нежелательные и даже вредные. Препарат становится менее избирательно действующим независимо от того, относится вещество к природным соединениям или синтезировано.
D.P.McDonnell, B.L.Wagner
(перевод Малярской М.М.)
Введение
Биологическое действие эстрогенов и прогестерона на органы- мишени, такие как матка, молочные железы и ЦНС, хорошо известно (1). Однако обнаружение рецепторов к эстрогенам (Эр) и прогестерону (Пр) в тканях, не являющихся классическими мишенями, свидетельствует о том, что стероидные гормоны регулируют не только репродуктивные процессы. В частности, показано защитное влияние эстрогенов на костную ткань, препятствующее развитию остеопороза у женщин в постменопаузе (2). Заместительная гормональная терапия эстрогенами (ЗГТ), помимо влияния на кости, снижает риск развития сердечно-сосудистых расстройств, обычно повышающийся после менопаузы (3). В настоящее время неизвестно, каким образом половые стероидные гормоны реализуют свое действие в этих системах организма. Повышенный интерес к изучению молекулярных и генетических основ строения Эр и Пр связан с разработкой новых модуляторов этих рецепторов, которые предполагается использовать при лечении сердечно-сосудистых заболеваний, остеопороза (ОП), а также репродуктивных нарушений и гормонозависимого рака.
Рецепторы к эстрогенам и прогестерону и их генетический контроль
Биологическое действие эстрогенов и прогестерона реализуется через высокоаффинные ядерные рецепторы в клетках-мишенях. Альфа-эстрогеновый рецептор (Эр) представляет собой простой белок с молекулярной массой 25 кД (4). Недавно была открыта бета-форма Эр, представленость этих форм в тканях-мишенях различается (глава 11). Существующие различия между подтипами Эр, заключающиеся в выраженности аффинитета к лигандам и распределении в тканях, могут объяснять селективное действие агонистов и антагонистов Эр в различных тканях. Открыты и разновидности мРНК, потенциально кодирующие синтез других подтипов Эр, однако биологическая значимость этих возможных подтипов пока не изучена (5). Пр также существуют в виде двух различных форм — Пр-А и Пр-В, 94 и 114 кД, соответственно (6). Обе формы кодируются одним геном, путем альтернативной инициации транскрипции с разных промоторов (7).
Изучаемые рецепторы относятся к большому семейству ядерных белков, опосредующих биологическое действие стероидных гормонов, гормонов щитовидной железы, витаминов Д и А (8). Анализ нуклеотидной последовательности ДНК, кодирующей синтез этих рецепторов, а также изучение биологической активности изоформ рецепторных белков, полученных в результате мутаций гена, показали их сходную структуру (8, 10). Домен, связывающий лиганд, находится на карбоксильном конце полипептидной цепи и состоит из 300 аминокислот, а домен, связывающий ДНК, состоит из 66-68 аминокислот. Этот домен, находящийся на Nh3-конце белка, наиболее вариабелен по строению, и именно он вовлечен в регуляцию транскрипции (7, 11-15). Предполагается, что последовательность, активирующая этот домен (TAF), для Эр и Пр-А находится в одном месте, а для Пр-В существует дополнительная последовательность (BUS) (17, 18).
Регуляция транскрипции эстрогеновыми и прогестероновыми рецепторами
Общий механизм рействия рецепторов к стероидным гормонам сходен (19, 20). В отсутствие гормонов рецепторы находятся в ядре клеток-мишеней в латентной форме, связанные с высокомолекулярным белком теплового шока 90 (hsp90), hsp 72, hsp 59 и возможно другими белками (21-23), рис. 12.1. Точная стехиометрия комплекса рецептор-белки теплового шока до конца не изучена, однако считается, что в состав одного олигомерного комплекса входит только одна молекула рецептора (24). Роль белков теплового шока в регуляции активности рецепторов неизвестна, они могут участвовать в поддержании рецептора в неактивном состоянии, пока нет взаимодействия с гормонами (25-27). Предполагается, что обратимое связывание со стероидным лигандом приводит к конформационным изменениям молекулы рецептора, высвобождающим его из комплекса с белками теплового шока (23). Несмотря на то, что все рецепторы к стероидным гормонам связаны с hsp 90, похоже, что роль этого белка в регуляции биологической активности разных рецепторов разная. Например, показано, что hsp 90 участвует в образовании гормон-связывающей активной формы рецептора к глюкокортикоидам (8), но не участвует в образовании активной формы связывающего домена Эр (29). Очень мало известно о роли других белков теплового щока, связанных с неактивным рецептором (21, 30).
Инициация передачи сигнала прогестероновым рецептором
Взаимодействие молекулы прогестерона с Пр инициирует каскад передачи сигнала путем отсоединения белков теплового шока (31) и образования стабильного рецепторного димера. В клетках, одновременно экспрессирующих Пр-А и Пр-В, существует 3 различных типа димеров (А:А, А:В, В:В). Относительная плотность каждого димера пропорциональна уровню экспрессии Пр-А и Пр-В (31, 32). Активированный рецепторный димер способен взаимодействовать со специфическими элементами ответа на прогестерон (PRE), расположенными в промоторе регулируемого гена (33). Помимо образования рецепторного димера, взаимодействие рецептора с гормоном повышает уровень фосфорилирования Пр (34-36). Активация фосфорилирования происходит двумя невзаимосвязанными способами: путем отделения белков теплового шока и взаимодействия рецептора с ДНК (35, 36). Специфическое значения этого фосфорилирования до сих пор неизвестно.
Инициация передачи сигнала эстрогеновым рецептором
Взаимодействие Эр со специфической последовательностью ДНК в регуляторном участке гена-мишени — конечный итог в цепи передачи сигнала. Однако роль гормонов в осуществлении этого взаимодействия противоречива (37). В то время как считалось доказанным, что гормон необходим для взаимодействия Эр с ДНК в интактных клетках (37-40), анализ Эр-ДНК взаимодействия in vitro показал отсутсвие такой необходимости (41). Дальнейшие исследования показали, что перенос гена, экспрессирующего Эр, в клетки HeLa, в отсутствие лиганда было достаточным для транскрипции регулируемого гена (42). Возможно, что этот тип лиганд-независимой активации является следствием избыточной экспрессии рецепторов.
Рис. 12.1. Механизм действия стероидных гормонов
Стероидные гормоны реализуют свой эффект путем транскрипции генов с помощью специфических внутриклеточных рецепторных белков. Изученные генетические и биохимические процессы позволяют предположить, что передача сигнала в ядро осуществляется серией последовательных шагов. Гормон заходит в клетку пассивно, находит свой рецепторв (SR) в комплексе с белками теплового шока (hsp). Связывание лиганда инициирует каскад реакций, включающих фосфорилирование, димеризацию, ядерную транслокацию, взаимодействие со специфическими элементами ответа в ДНК (SRE) и активации адапторного белка, позволяющего стероидному рецептору активно взаимодействовать с аппаратом транскрипции (GTA). Конечным итогом клеточных и ядерных взаимодействий является активирующее влияние на РНК-полимеразу и усиление трранскрипции регулируемого гена.
Эр и Пр как факторы транскрипции
В последнее время предпринимаются шаги в изучении механизма регуляции транскрипции РНК-полимеразой II (43). Транскрипция в эукариотической клетке является сложным комплексом взаимодействий между белками, связанными в специфическом участке ДНК, и промотором. Промотерные последовательности расположены в начале транскрибируемого участка (core promoter) и в обе стороны от него (enhancer). Core promoter, необходимый для начала транскрипции, состоит инициального комплекса, расположенного +1 и комплекса ТАТА, расположенного -25 (44). Эти элементы промотора при соответствующей стимуляции способствуют прикреплению РНК-полимеразы к промотору. Активированные Эр и Пр участвуют в транскрипции, связываясь с промоторными последовательностями регулируемого гена (45). Точный механизм влияния Эр и Пр на РНК-полимеразу II неизвестен. Показано in vitro потенцирование транскрипции с помощью активного Пр путем стабилизирования инициального комплекса (46). Показано взаимодействие Эр и Пр с общим фактором транскрипции (TFIIB) (47), что свидетельствует о прямом влиянии рецепторов на аппарат транскрипции. Кроме прямого влияния, гормон-активированный рецептор может влиять на транскрипцию путем медиаторных белков. Похоже, что эти белки по-разному экспрессируются в клетках-мишенях, и в связи с этим их уровень экспрессии может влиять на чувствительность клеток к гормонам; также они влияют на процесс распознавания агонистов и антагонистов рецепторов (16, 48). Недавно были выделены и клонированы медиаторные белки, селективно взаимодействующие с Эр и Пр, активированными именно агонистами (49-52).
Антагонисты рецепторов стероидных гормонов
Существует две модели действия антагонистов стероидных рецепторов. Первая — конкурентное взаимодействие, блокирующее центр связывания с эндогенными лигандами. При этом рецептор остается в неактивном, латентном состоянии (рис. 12.2.А). Вторая модель — псевдоагонизм, подражание естественным агонистам, однако после связывания с псевдоагонистом рецептор начинает подавлять транскрипцию. В результате этого взаимодействия «неактивный» рецептор (рис. 12.2В) может конкурировать с активированным агонистом рецептором за связывание с ДНК. Фармакологические влияния двух типов антагонистов различаются (53). Вторая модель лучше объясняет механизм действия большинства известных антагонистов стероидных рецепторов.
Рис. 12.2. Возможный механизм действия антагонистов рецепторов к стероидным гормонам
В отсутствие гормона рецептор находится в неактивном состоянии в ядре клетки-мишени. Гормон свзяывается с рецептором, это инициирует каскад реакций, приводящих в конечном итоге к изменению скорости транскрипции регулируемого гена. Существует как минимум два типа антагонистов стероидных рецепторов. Первый класс конкурентно взаимодействует с рецептором и блокирует действие агонистов. Этот пассивный тип ингибирования удерживает рецептор в латентном состоянии в то время как антагонист физически связан с ним. Альтернативой служит второй, активный тип ингибирования. Такие антагонисты действуют как псевдоагонисты, подражая некоторым их эффектам. Такие антагонисты переводят Пр из латентной формы в т.н. неактивную, которая обладает дополнительной ингибиторной активностью, конкурируя с активной формой, связанной с гормоном, за связывание с промоторными участками ДНК, и блокируя аппарат транскрипции. Все кроме одного существующие сегодня антигормоны относятся к активным ингибиторам или псевдоагонистам.
Антагонисты прогестероновых рецепторов
Существующие антипрогестины прямо взаимодействуют с гормон-связывающим доменом Пр, конкурентно блокируя связывание прогестерона. Кроме одного исключения (ZK98299; Onapristone), взаимодействие рецептора с антигормонами достаточно для его связывания с регулируемой последовательностью ДНК (54, 55). Механизм действия ZK98299 отличается тем, что он связывается с рецептором, но не приводит к образованию высокоаффинного комплекса Пр-ДНК in vitro. Одно из возможных объяснений — ZK98299 предотвращает димеризацию рецептора, необходимый шаг для связывания ДНК. Т.о. антипрогестины можно разделить на два типа: тип 1 — предотвращающие связывание с ДНК (онапристон) и тип 2 — способствующие связыванию (Рис. 12.3). В отличие от онапристона (тип 1), антипрогестины 2 типа проявляют частичную активность агонистов в соответствующих условиях. Однако в последнее время появляются доказательства того, что различия между типами антипрогестинов касаются не столько строения, сколько аффинитета. Это соответствует данным лаборатории Milgrom, сотрудники которой показали, что in vivo онапристон в насыщающей концентрации оказывает действие идентичное антипрогестинам 2 класса (58). Однако на сегодняшний день сохраняется классификация антипрогестинов в зависимости от того, предотвращают (тип 1) или активируют они взаимодействие рецептора с ДНК in vitro (с использованием номенклатуры Klein-Hipass, 54).
Хотя и агонисты и антагонисты взаимодействуют с лиганд-связывающим доменом, было показано с помощью мутагенеза рецепторов, что связывающие последовательности для агонистов и антагонистов хоть и перекрываются, но все-таки различны (59, 60). Идентицифицирован мутантный Пр (Пр-Up-1), в котором отсутствует 54-я с СООН-конца аминокислота; с этим мутантным рецептором могут связываться антагонисты и не могут агонисты. Более того, при связывании с этим рецептором антипрогестин Ру486 проявляет себя как агонист. В серии экспериментов Benhamou et al идентифицировали аминокислоты в лиганд-связывающем домене Пр, изменение которых нарушало связывание антагонистов, но не агонистов (59). Эти данные демонстрируют, что последовательности, необходимые для связывания прогестерона и Ру486 различны. Второй вывод заключается в том, что карбоксильный конец Пр может быть частью функционального комплекса, который в отсутствие гормона поддерживает рецептор в неактивном состоянии. Мы продолжили эти исследования и продемонстрировали, что существует третий класс лигандов, т.н. смешанные агонисты, взаимодействующие с лиганд-связывающим доменом по механизму, отличающемуся и от агонистов и от антагонистов (61). Т.о. мы пришли к выводу о том, что биологическое действие лиганда Пр зависит от механизма его взаимодействия с рецептором (48, 60, 62).
Важным ключом к пониманию того, каким образом в клетке происходит различение лигандов Пр, являются результаты исследования Allan et al (62). Авторы, изучая строение синтезированного in vitro Пр при помощи частичного протеолиза продемонстрировали, что прогестерон и Ру486 вызывают различные конформационные изменения рецепторного белка. Используя специфические моноклональные антитела, исследователи показали, что конформационные изменения происходят в карбоксильном конце рецептора (60, 62). Wagner et al, продолжая подобные эксперименты, показали, что смешанные агонисты Пр вызывают изменения структуры рецептора, отличающиеся от таковых, вызываемых агонистами и антагонистами (61). Была отмечена связь между структурой лиганд-рецепторного комплекса и конечным биолоическим действием. Эти исследования позволяют предположить возможность синтеза специфичных Пр-модуляторов, вызывающих различные конформации рецепторного белка и следовательно различные биологические эффекты.
Используя информацию, полученную из вышеописанных исследований, мы сформулировали рабочую гипотезу, объясняющую механизм, с помощью которого Пр различают связывающиеся с ними лиганды (рис. 12.4). Мы предполагаем, что карбоксильный конец Пр служит репрессором транскрипции. Взаимодействие с Пр агониста, антагониста или смешанного агониста приводит к различным конформационным изменениям рецепторного белка, которые в конечном итоге приводят к отщеплению белков теплового шока. Однако только агонист и в меньшей степени частичный агонист вызывают конформации, которые могут преодолеть ингибиторное влияние концевой части домена и привести к продуктивному взаимодействию рецептора с аппаратом транскрипции. Подтверждение этой гипотезы требует кристаллографической информации, однако наши наблюдения уже показали, что различные лиганды взаимодействуют с Пр различными путями и приводят к различным биологическим эффектам.
Рис. 12.5. Механизм действия агонистов и антагонистов Пр
Основываясь на опубликованных данных и дополнительных данных, представленных в настоящем обзоре, мы предположили следующую рабочую гипотезу, объясняющую клеточный механизм различения агонистов и антагонистов Пр. В свободном состоянии Пр неактивны и взаимодействуют с репрессорными белками. Агонисты связываются с карбоксильным концом рецепторного белка и вызывают структурные изменения, активирующие рецептор, возможно путем удаления репрессорного белка и/или активации домена, связывающегося с ДНК. Антагонисты, связывающиеся с NH 2 -концом гормон-связывающего домена, вызывают другое изменение конформации рецепторного белка, при котором карбоксильный конец не повернут к рецепторной части. Изменения, вызываемые антагонистами, могут быть недостаточны для удаления репрессорного белка или активации доменов связывания с ДНК. Смешанные агонисты могут связываться и с карбоксильной и с аминогруппой гормон-связывающего домена, вызывая новую конформацию, позволяющую рецептору проявлять некоторую активность, но не полноценно, например путем продолжающейся, но менее стабильной связи с репрессором.
Антагонисты эстрогеновых рецепторов
Некоторые из наиболее часто использующихся антиэстрогенов, как в терапии, так и в экспериментальной эндокринологии, показаны на рис. 2.5. По результатам экспериментов in vitro и in vivo, эти вещества были разделены на разные категории. Существуют как стероидные (ICI164,384), так и нестероидные антиэстрогены (тамоксифен) (16, 48, 63, 64). Антиэстроген ICI164,384 является стероидом и вначале создавался как активный лиганд Эр. Было показано, что добавление длинного алкильного радикала в положении 7альфа к стероидному ядру приводит к антиэстрогенной активности соединения. Свойства этого класса антиэстрогенов изучены достаточно широко, поскольку они являются первым классом, лишенным частичной эстрогенной активности. Другой класс широко используемых антиэстрогенов — трифенилэтилены (тамоксифен) и производные бензотиофена (ралоксифен). Эти вещества уникальны тем, что в большинстве тканей они являются антагонистами Эр, а в некоторых клетках и при определенных условиях проявляют частичную активность агонистов. В частности, эти вещества проявляют антиэстрогенную активность в отношении молочных желез и эстрогенную — в отношении костей и сердечно-сосудистой системы. Молекулярные основы этого феномена достаточно изучены и изложены в литературе (16, 41, 47, 48, 56, 65).
Стероидный антиэстроген ICI164,384 проявляет исключительно антиэстрогенную активность как in vitro, так in vivo. Предполагается, что ICI164, 384 блокирует функцию Эр, нарушая димеризацию (41, 63, 66). Однако данные об изменении эффективности связывания Эр с ДНК после взаимодействия с ICI164,384 противоречивы. Было показано, что стабильность Эр-димеров может колебаться в зависимости от клеточной локализации Эр (67) и от условий эксперимента, используемых для определения взаимодействия Эр-димеров с ДНК. Эти различия могут объяснить, почему некоторые исследователи не могут обнаружить какие-либо влияния ICI164,384 на взаимодействия Эр с ДНК (68). В отличие от ICI164,384, производные бензотиофена (ралоксифен) и трифенилэтилена (тамоксифен) способствуют взаимодействию Эр с ДНК. В большинстве случаев эта активность приводит к антиэстрогенной активности (16, 48, 65). Однако относительная выраженность свойств этих веществ как агонистов и антагонистов зависит от окружающих условий. На основании серии in vitro-экспериментов были сформулированы молекулярные критерии, позволяющие различать агонистов Эр и частичных агонистов, а также дополнительно классифицировать известные антагонисты Эр на три функциональных категории. Эффекты известных агонистов и антагонистов позволяет предположить, что эта классификация отражает различные изменения Эр, вызыванные лигандами. Схема этой классификации отражена на рис. 12.6. Было предположено, что Эр существует в состоянии, среднем между активной и неактивной формой, причем в отсутствии лиганда преобладает неактивная конформация. Взаимодействие Эр с 17-бета-эстрадиолом стабилизирует комплекс в конформации, активирующей транскрипцию. Относительная про-/антиэстрогенная активность других модуляторов Эр определяется промежуточными конформациями. Адаптируя принцип Klein-Hitpass et al, первоначально предложенный для классификации антипрогестинов, предложено назвать вещества, препятствующие взаимодействию Эр с ДНК, антиэстрогенами 1 типа (54). В отличие от 1 класса антипрогестинов, антиэстрогены 1 типа на сегодняшний день не идентифицированы. Основываясь на этой номенклатуре, предложено ICI164,384 отнести к антиэстрогенам 2 типа, вызывающим конформацию рецептора, наиболее близкую к неактивной. Ралоксифен, который может проявлять себя как антагонист и частичный агонист, в зависимости от условий, представляет собой 3 тип антиэстрогенов. Наконец, 4-ОН-тамоксифен представляет 4 тип антиэстрогенов, стабилизирующий Эр в наиболее активной конформации (16, 48). Со временем возможен синтез новых типов антагонистов Эр.
Рис. 12.6. Гормоны и антигормоны, стабилизирующие различные конформационные состояния Эр.
Эр существует в клетках в различных конформациях, представляющих неактивное, активное и ряд промежуточных состояний. Лиганды проявляют свои биологические свойства, стабилизируя Эр в одном из его конформационных состояний. Основываясь на номенклатуре Klein-Hitpass (54) и результатах наших исследований, мы подразделили известные антиэстрогены на различные классы, представленные ICI164,384 (тип 2), ралоксифеном (тип 3), 4-гидрокситамоксифеном (тип 4), соответственно. Хотя мы не можем продемонстрировать существование антиэстрогенов 1 типа (предотвращающих взаимодействие рецептора с ДНК), теоретически возможно, что эти вещества будут открыты.
Характеристики агонистов
Агонисты могут быть эндогенными (например, гормоны и нейротрансмиттеры) и экзогенными (лекарства). Эндогенные агонисты в норме производятся внутри организма и опосредуют функцию рецептора. К примеру, дофамин является эндогенным агонистом дофаминовых рецепторов .
Физиологическим агонистом называется вещество, вызывающее аналогичный отклик, но действующее на иной рецептор.
Спектр эффектов
Спектр эффектов агонистов
Агонисты различаются по силе и направлению физиологического ответа, вызываемого ими. Данная классификация не связана с аффинностью лигандов и опирается лишь на величину отклика рецептора.
Суперагонист — соединение, способное вызывать более сильный физиологический ответ, чем эндогенный агонист. Полный агонист — соединение, вызывающее такой же отклик, как эндогенный агонист (например, изопреналин , агонист β-адренорецепторов). В случае меньшего отклика соединение называют частичным агонистом (например, арипипразол — частичный агонист дофаминовых и серотониновых рецепторов).
В случае, если у рецептора имеется базальная (конститутивная) активность, некоторые вещества — обратные агонисты — могут уменьшать её. В частности, обратные агонисты рецепторов ГАМК A обладают анксиогенным или спазмогенным действием, однако могут усиливать когнитивные способности .
Механизм
Если для активации рецептора требуется взаимодействие с несколькими различными молекулами, последние называются коагонистами. В качестве примера можно привести NMDA-рецепторы , активирующиеся при одновременном связывании глутамата и глицина .
Необратимым агонист называют в случае, если после связывания с ним рецептор становится постоянно активированным. В данном случае не имеет значения, образует ли лиганд ковалентную связь с рецептором либо взаимодействие является нековалентным, но чрезвычайно термодинамически выгодным.
Селективность
Селективным агонист называют в том случае, если он активирует лишь один конкретный рецептор либо подтип рецепторов. Степень селективности может различаться: дофамин активирует рецепторы пяти различных подтипов, но не активирует серотониновые рецепторы . В настоящее время встречаются экспериментальные подтверждения возможности различного взаимодействия одних и тех же лигандов с одними и теми же рецепторами: в зависимости от условий одно и то же вещество может быть полным агонистом, антагонистом или обратным агонистом.
Активность
Примечания
Wikimedia Foundation . 2010 .
Синонимы :Смотреть что такое «Агонист» в других словарях:
— (этим. см. пред. слово). Боец. Словарь иностранных слов, вошедших в состав русского языка. Чудинов А.Н., 1910. АГОНИСТ греч. agonistes, от agon, борьба. Противник, гонитель мнений. Объяснение 25000 иностранных слов, вошедших в употребление в… … Словарь иностранных слов русского языка
Сущ., кол во синонимов: 3 боец (39) гонитель (5) противник (26) Словарь синонимов ASIS … Словарь синонимов
агонист — Небольшие белки или органические молекулы, связывающиеся с определенными клеточными белками, которые являются рецепторами, вызывают их конформационные изменения, что усиливает действие горомонов, медиаторов и др… … Справочник технического переводчика
АГОНИСТ — 1. Мышца, которая сокращается и действует в противоположном направлении по сравнению с другой мышцей, антагонистом; при сгибании локтя, например, бицепс – агонист, а трицепс – антагонист. См. мышцы антагонисты. 2. Любое лекарственное средство,… … Толковый словарь по психологии
агонист — (грч agonistes) кај старите Грци борец, мегданџија, натпреварувач во витешки игри … Macedonian dictionary
АГОНИСТ — (agonist) 1. Prime mover мышца, за счет сокращения которой происходит определенное движение той или иной части тела. Сокращение мышцы агониста сопровождается расслаблением противодействующей ей мышцы антагониста. 2. Лекарственный препарат или… … Толковый словарь по медицине
Рецепторы (от лат. recipere — получать) представляют собой биологические макромолекулы, которые предназначены для связывания с эндогенными лигандами (нейротрансмиттерами, гормонами, факторами роста). Рецепторы могут взаимодействовать также с экзогенными биологически активными веществами, в т.ч. и с лекарственными.
При взаимодействии лекарственного вещества с рецептором развивается цепь биохимических превращений, конечным итогом которых является фармакологический эффект.
Выделяют четыре типа рецепторов:
1. Рецепторы, осуществляющие прямой контроль функции эффекторного фермента. Они связаны с плазматической мембраной клеток, фосфорилируют белки клеток и изменяют их активность. По такому принципу устроены рецепторы к инсулину, лимфокинам, эпидермальному и тромбоцитарному факторам роста.
2. Рецепторы, осуществляющие контроль за функцией ионных каналов. Рецепторы ионных каналов обеспечивают проницаемость мембран для ионов. Н-холинорецепторы, рецепторы глутаминовой и аспарагиновой кислот увеличивают проницаемость мембран для ионов + + 2+
Na , K , Ca , вызывая деполяризацию и возбуждение функции клеток. ГАМКА-рецепторы, глициновые рецепторы увеличивают проницаемость — мембран для Cl , вызывая гиперполяризацию и торможение функции клеток.
3. Рецепторы, ассоциированные с G-белками. При возбуждении этих рецепторов влияние на активность внутриклеточных ферментов опосредуется через G-белки. Изменяя кинетику ионных каналов и 2+ синтез вторичных мессенджеров (цАМФ, цГМФ, ИФ3, ДАГ, Са), G-белки регулируют активность протеинкиназ, которые обеспечивают внутриклеточное фосфорилирование важных регуляторных белков и развитие разнообразных эффектов. К числу таких рецепторов
относятся рецепторы для полипептидных гормонов и медиаторов (м-холинорецепторы, адренорецепторы, гистаминовые рецепторы). Рецепторы 1-3 типов локализованы на цитоплазматической мембране.
4. Рецепторы — регуляторы транскрипции ДНК. Эти рецепторы являются внутриклеточными и представляют собой растворимые цитозольные или ядерные белки. С такими рецепторами взаимодействуют стероидные и тиреоидные гормоны. Функция рецепторов — активация или ингибирование транскрипции генов.
Рецепторы, обеспечивающие проявление действия определенных веществ, называют специфическими.
Вещества, которые при взаимодействии со специфическими рецепторами вызывают в них изменения, приводящие к биологическому эффекту, называют агонистами. Стимулирующее действие агониста на рецепторы может приводить к активации или угнетению функции клетки. Если агонист, взаимодействуя с рецепторами, вызывает максимальный эффект, то это полный агонист. В отличие от последнего частичные агонисты при взаимодействии с теми же рецепторами не вызывают максимального эффекта.
Вещества, связывающиеся с рецепторами, но не вызывающие их стимуляции, называют антагонистами. Их внутренняя активность равна нулю. Их фармакологические эффекты обусловлены антагонизмом с эндогенными лигандами (медиаторами, гормонами), а также с экзогенными веществами-агонистами. Если они оккупируют те же рецепторы, с которыми взаимодействуют агонисты, то речь идет о конкурентных антагонистах; если другие участки макромолекулы, не относящиеся к специфическому рецептору, но взаимосвязанные с ним, то говорят о неконкурентных антагонистах.
Фармакодинамика включает понятия о фармакологических эффектах, локализации действия и механизмах действия ЛВ (т.е. представления о том, как, где и каким образом ЛВ действуют в организме). К фармакодинамике относится также понятие о видах действия ЛВ.
2.1. ФАРМАКОЛОГИЧЕСКИЕ ЭФФЕКТЫ, ЛОКАЛИЗАЦИЯ И МЕХАНИЗМЫ ДЕЙСТВИЯ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ
Фармакологические эффекты — изменения функ- ции органов и систем организма, вызываемые ЛВ. К фармакологическим эффектам ЛВ относятся, например, повышение частоты сердечных сокращений, снижение артериального давления, повышение порога болевой чувствительности, снижение температуры тела, увеличение продолжительности сна, устранение бреда и галлюцинаций и т.п. Каждое вещество, как правило, вызывает ряд определенных, характерных для него фармакологических эффектов. При этом одни фармакологические эффекты ЛВ являются полезными — благодаря им ЛВ применяют в медицинской практике (основные эффекты),
а другие не используются и, более того, являются нежелательными (побочные эффекты).
Для многих веществ известны места их преимущественного действия в организме — т.е. локализация действия. Одни вещества преимущественно действуют на определенные структуры головного мозга (противопаркинсонические, антипсихотические средства), другие в основном действуют на сердце (сердечные гликозиды).
Благодаря современным методическим приемам, можно определить локализацию действия веществ не только на системном и органном, но на клеточном и молекулярном уровнях. Например, сердечные гли- козиды действуют на сердце (органный уровень), на кардиомиоциты (клеточный уровень), на Na + -, К + -АТФазу мембран кардиомиоцитов (молекулярный уровень).
Одни и те же фармакологические эффекты могут быть вызваны различными способами. Так, есть вещества, которые вызывают сни- жение артериального давления, уменьшая синтез ангиотензина II (ингибиторы АПФ), или блокируя поступление Са 2+ в гладкомышечные клетки (блокаторы потенциалзависимых кальциевых каналов) или уменьшая выделение норадреналина из окончаний симпатических нервов (симпатолитики). Способы, с помощью которых ЛВ вызывают фармакологические эффекты, определяются как м е х а н и з — мы действия.
Фармакологические эффекты большинства ЛВ вызываются их действием на определенные молекулярные субстраты, так называемые «мишени».
К основным молекулярным «мишеням» для ЛВ относятся рецепторы, ионные каналы, ферменты, транспортные системы.
Рецепторы
А. Свойства и виды рецепторов. Взаимодействие рецепторов с ферментами и ионными каналами
Рецепторы представляют собой функционально активные макромолекулы или их фрагменты (в основном это белковые молекулы — липопротеины, гликопротеины, нуклеопротеины и др.). При взаимодействии веществ (лигандов) с рецепторами возникает цепь биохимических реакций, приводящая к развитию определенных
фармакологических эффектов. Рецепторы служат мишенями для эндогенных лигандов (нейромедиаторов, гормонов, других эндоген- ных биологически активных веществ), но могут взаимодействовать и с экзогенными биологически активными веществами, в том числе с ЛВ. Рецепторы взаимодействуют только с определенными веществами (имеющими определенную химическую структуру и пространственную ориентацию), т.е. обладают избирательностью, поэтому их называют специфическими рецепторами.
Рецепторы не являются стабильными, постоянно существующими структурами клеток. Количество их может увеличиваться вследствие преобладания синтеза рецепторных белков или уменьшаться вследствие превалирования процесса их деградации. Кроме того, рецепторы могут терять свою функциональную активность (десенситизация), вследствие чего при взаимодействии рецептора с лигандом не возникают биохимические реакции, приводящие к фармакологическому эффекту. Все эти процессы регулируются концентрацией лиганда и длительностью его воздействия на рецепторы. При длительном воздействии лиганда развивается десенситизация рецепторов и/или снижение их количества (down-регуляция), и, наоборот, отсутствие лиганда (или снижение его концентрации) приводит к увеличению количества рецепторов (up-регуляция).
Рецепторы могут находиться в мембране клеток (мембранные рецепторы) или внутри клеток — в цитоплазме или ядре (внутрикле- точные рецепторы) (рис. 2-1).
Мембранные рецепторы. В мембранных рецепторах выделяют внеклеточный и внутриклеточный домены. На внеклеточном домене имеются места связывания для лигандов (веществ, взаимодействующих с рецепторами). Внутриклеточные домены взаимодействуют с эффекторными белками (ферментами или ионными каналами) или сами обладают ферментативной активностью.
Известны три вида мембранных рецепторов.
1. Рецепторы, непосредственно сопряженные с ферментами. Поскольку внутриклеточный домен этих рецепторов проявляет ферментативную активность, их называют также рецепторами-ферментами, или каталитическими рецепторами. Большинство рецепторов этой группы обладает тирозинкиназной активностью. При связывании рецептора с веществом происходит активация тирозинкиназы, которая фосфорилирует внутриклеточные белки и таким образом изменяет их активность. К этим рецепторам относят рецепторы для инсулина, некоторых факторов роста и цитокинов. Известны рецепторы, непосредственно связанные с гуанилатциклазой (при воздействии на них предсердного натрийуретического фактора, происходит активация гуанилатциклазы, и в клетках увеличивается содержание циклического гуанозинмонофосфата).
2. Рецепторы, непосредственно сопряженные с ионными каналами, состоят из нескольких субъединиц, которые пронизывают клеточную мембрану и формируют ионный канал. При связывании вещества с внеклеточным доменом рецептора ионные каналы открываются, в результате изменяется проницаемость клеточных мембран для различных ионов. К таким рецепторам относятся Н-холинорецепторы, рецепторы к гамма-аминомасляной кислоте (ГАМК), относящиеся к подтипу А, глициновые рецепторы, глутаматные рецепторы.
Н-холинорецептор состоит из пяти субъединиц, пронизывающих клеточную мембрану. При связывании двух молекул ацетилхолина с двумя α-субъединицами рецептора открывается натриевый канал и ионы натрия поступают в клетку, вызывая деполяризацию клеточной мембраны (в скелетных мышцах это приводит к мышечному сокращению).
ГАМК А -рецепторы непосредственно сопряжены с хлорными каналами. При взаимодействии рецепторов с ГАМК хлорные каналы открываются и ионы хлора поступают в клетку, вызывая
гиперполяризацию клеточной мембраны (это приводит к усилению тормозных процессов в ЦНС). Таким же образом функцио- нируют глициновые рецепторы. 3. Рецепторы, взаимодействующие с G-белками. Эти рецепторы взаимодействуют с ферментами и ионными каналами клеток через белки-посредники (G-белки — гуанозинтрифосфат (GTP)-связывающие белки). При действии вещества на рецептор α-субъединица G-белка связывается с гуанозинтрифосфатом. При этом комплекс G-белок-гуанозинтрифосфат вступает во взаимодействие с ферментами или ионными каналами. Как правило, один рецептор сопряжен с несколькими G-белками, а каждый G-белок может одновременно взаимодействовать с несколькими молекулами ферментов или несколькими ионными каналами. В результате такого взаимодействия происходит усиление (амплификация) эффекта.
Хорошо изучено взаимодействие G-белков с аденилатциклазой и фосфолипазой С.
Аденилатциклаза — мембраносвязанный фермент, гидролизующий АТФ. В результате гидролиза АТФ образуется циклический аденозинмонофосфат (цАМФ), который активирует цАМФ-зависимые протеинкиназы, фосфорилирующие клеточные белки. При этом изменяется активность белков и регулируемых ими процессов. По влиянию на активность аденилатциклазы G-белки подразделяются на G s -белки, стимулирующие аденилатциклазу, и G i -белки, ингибирующие этот фермент. Примером рецепторов, взаимодействующих с G s -белками, являются β 1 -адренорецепторы (опосредуют стимулирующее влияние на сердце симпатической иннервации), а рецепторов, взаимодействующих с G i -белками — М 2 -холинорецепторы (опосредуют тормозное влияние на сердце парасимпатической иннервации). Эти рецепторы локализованы в мембране кардиомиоцитов.
При стимуляции β 1 -адренорецепторов повышается активность аденилатциклазы и увеличивается содержание цАМФ в кардиомиоцитах. В результате активируется протеинкиназа, которая фосфорилирует кальциевые каналы мембран кардиомиоцитов. Через эти каналы ионы кальция поступают в клетку. Вход Са 2+ в клетку увеличивается, что приводит к повышению автоматизма синусного узла и увеличению частоты сердечных сокращений. Внутриклеточные эффекты противоположной направленности развиваются при стимуляции М 2 -холинорецепторов кардиомиоцитов, в результате происходит снижение автоматизма синусного узла и частоты сердечных сокращений.
С фосфолипазой С взаимодействуют G q -белки, вызывая ее активацию. Примером рецепторов, сопряженных с G q -белками, являются а г адренорецепторы гладкомышечных клеток сосудов (опосредующие влияние на сосуды симпатической иннервации). При стимуляции этих рецепторов повышается активность фосфолипазы С. Фосфолипаза С гидролизует фосфатидилинозитол-4,5-дифосфат клеточных мембран с образованием гидрофильного вещества инозитол-1,4,5-трифосфа- та, который взаимодействует с кальциевыми каналами саркоплазматического ретикулума клетки и вызывает высвобождение Са 2+ в цитоплазму. При повышении концентрации Са 2+ в цитоплазме гладкомышечных клеток увеличивается скорость образования комплекса Са 2+ -кальмодулин, который активирует киназу легких цепей миозина. Этот фермент фосфорилирует легкие цепи миозина, в результате чего облегчается взаимодействие актина с миозином, и происходит сокращение гладких мышц сосудов.
К рецепторам, взаимодействующим с G-белками, относятся также дофаминовые рецепторы, некоторые подтипы серотониновых (5-НТ) рецепторов, опиоидные рецепторы, гистаминовые рецепторы, рецепторы для большинства пептидных гормонов и др.
Внутриклеточные рецепторы представляют собой растворимые цитозольные или ядерные белки, которые опосредуют регулирующее действие веществ на транскрипцию ДНК. Лигандами внутриклеточных рецепторов являются липофильные вещества (стероидные и тиреоидные гормоны, витамины А, Д).
Взаимодействие лиганда (например, глюкокортикоидов) с цитозольными рецепторами вызывает их конформационное изменение, в результате комплекс вещество-рецептор перемещается в ядро клетки, где связывается с определенными участками молекулы ДНК. Происходит изменение (активация или репрессия) транскрипции генов, кодирующих синтез различных функционально активных белков (ферментов, цитокинов и т.д.). Увеличение (или уменьшение) синтеза ферментов и других белков приводит к изменению биохи- мических процессов в клетке и возникновению фармакологических эффектов. Так, глюкокортикоиды, активируя гены, ответственные за синтез ферментов глюконеогенеза, стимулируют синтез глюкозы, что способствует развитию гипергликемии. В результате репрессии генов, кодирующих синтез цитокинов, молекул межклеточной адгезии, циклооксигеназы, глюкокортикоиды оказывают иммунодепрессивное и противовоспалительное действие. Фармакологические
эффекты веществ при их взаимодействии с внутриклеточными рецепторами развиваются медленно (в течение нескольких часов и даже суток).
Взаимодействие с ядерными рецепторами характерно для тиреоидных гормонов, витаминов А (ретиноидов) и Д. Обнаружен новый подтип ядерных рецепторов — рецепторы, активируемые пролифераторами пероксисом. Эти рецепторы участвуют в регуляции липидного обмена и других метаболических процессов и являются мишенями для клофибрата (гиполипидемического препарата).
Б. Связывание вещества с рецептором. Понятие об аффинитете
Для того чтобы ЛВ подействовало на рецептор, оно должно с ним связаться. В результате образуется комплекс «вещество-рецептор». Образование подобного комплекса осуществляется с помощью межмолекулярных связей. Существует несколько видов таких связей.
Ковалентные связи — самый прочный вид межмолекулярных связей. Они образуются между двумя атомами за счет общей пары электронов. Ковалентные связи наиболее часто обеспечивают необратимое связывание веществ, однако они не характерны для взаимодействия ЛВ с рецепторами.
Ионные связи менее прочные, возникают между группировками, несущими разноименные заряды (электростатическое взаимо- действие).
Ион-дипольные и диполь-дипольные связи близки по характеру к ионным связям. В электронейтральных молекулах ЛВ, попадающих в электрическое поле клеточных мембран или находящихся в окружении ионов, происходит образование индуцированных диполей. Ионные и дипольные связи характерны для взаимодействия ЛВ с рецепторами.
Водородные связи играют весьма существенную роль во взаимодействии ЛВ с рецепторами. Атом водорода способен связывать атомы кислорода, азота, серы, галогенов. Водородные связи — слабые, для их образования необходимо, чтобы молекулы находились друг от друга на расстоянии не более 0,3 нм.
Ван-дер-ваальсовы связи — наиболее слабые связи, образуются между двумя любыми атомами, если они находятся на расстоянии не более 0,2 нм. При увеличении расстояния эти связи ослабевают.
Гидрофобные связи образуются при взаимодействии неполярных молекул в водной среде.
Для характеристики связывания вещества с рецептором используют термин aффинитет.
Аффинитет (от лат. affinis — родственный) — способность вещества связываться с рецептором, в результате чего происходит образование комплекса «вещество-рецептор». Кроме того, термин «аффинитет» используют для характеристики прочности связывания вещества с рецептором (т.е. продолжительности существования комплекса «вещество-рецептор»). Количественной мерой аффинитета как прочности связывания вещества с рецептором является константа диссоциации (К d).
Константа диссоциации равна концентрации вещества, при которой половина рецепторов в данной системе связана с веществом. Выражается этот показатель в молях/л (М). Между аффинитетом и константой диссоциации существует обратно пропорциональное соотношение: чем меньше К d , тем выше аффинитет. Например, если К d вещества А равна 10 -3 М, а К d вещества В равна 10 -10 М, аффинитет вещества В выше, чем аффинитет вещества А.
В. Внутренняя активность лекарственных веществ. Понятие об агонистах и антагонистах рецепторов
Вещества, которые обладают аффинитетом, могут иметь внутреннюю активность.
Внутренняя активность — способность вещества при взаимодействии с рецептором стимулировать его и таким образом вызывать определенные эффекты.
В зависимости от наличия внутренней активности ЛВ подразделяют на aгонисты и aнтагонисты рецепторов.
Агонисты (от греч. agonistes — соперник, agon — борьба) или миметики — вещества, обладающие аффинитетом и внутренней активностью. При взаимодействии со специфическими рецепторами они стимулируют их, т.е. вызывают изменения конформации рецеп- торов, в результате чего возникает цепь биохимических реакций и развиваются определенные фармакологические эффекты.
Полные агонисты, взаимодействуя с рецепторами, вызывают максимально возможный эффект (обладают максимальной внутренней активностью).
Частичные агонисты при взаимодействии с рецепторам вызывают эффект, меньший максимального (не обладают максимальной внутренней активностью).
Антагонисты (от греч. antagonisma — соперничество, anti — против, agon — борьба) — вещества, обладающие аффинитетом, но лишенные внутренней активности. Связываясь с рецепторами, они препятствуют действию на эти рецепторы эндогенных агонистов (нейромедиаторов, гормонов). Поэтому антагонисты также называют б л о к а т о р а м и рецепторов. Фармакологические эффекты антагонистов обусловлены устранением или ослаблением действия эндогенных агонистов данных рецепторов. При этом возникают эффекты, противоположные эффектам агонистов. Так, ацетилхолин вызывает брадикардию, а антагонист М-холинорецепторов атропин, устраняя действие ацетилхолина на сердце, повышает частоту сердечных сокра- щений.
Если антагонисты занимают те же места связывания, что и агонисты, они могут вытеснять друг друга из связи с рецепторами. Подобный вид антагонизма обозначают как конкурентный антагонизм, а антагонисты называют конкурентными антагониста- м и. Конкурентный антагонизм зависит от сравнительного аффинитета конкурирующих веществ к данному рецептору и их концентрации. В достаточно высоких концентрациях даже вещество с низким аффинитетом может вытеснить вещество с более высоким аффинитетом из связи с рецептором. Поэтому при конкурентном антагонизме эффект агониста может быть полностью восстановлен при увеличении его концентрации в среде. Конкурентный антагонизм часто используют для устранения токсических эффектов ЛВ.
Частичные антагонисты также могут конкурировать с полными агонистами за места связывания. Вытесняя полные агонисты из связи с рецепторами, частичные агонисты уменьшают их эффекты и поэтому в клинической практике могут быть использованы вместо антагонистов. Например, частичные агонисты β-адренорецепторов (пиндолол) так же, как антагонисты этих рецепторов (пропранолол, атенолол) применяют при лечении гипертонической болезни.
Неконкурентный антагонизм развивается, когда антагонист занимает так называемые аллостерические места связывания на рецепторах (участки макромолекулы, не являющиеся местами связывания агониста, но регулирующие активность рецепторов). Неконкурентные антагонисты изменяют конформацию рецепторов
таким образом, что они теряют способность взаимодействовать с агонистами. При этом увеличение концентрации агониста не может привести к полному восстановлению его эффекта. Неконкурентный анта- гонизм также имеет место при необратимом (ковалентном) связывании вещества с рецептором.
Некоторые ЛВ сочетают способность стимулировать один подтип рецепторов и блокировать другой. Такие вещества обозначают как агонистыантагонисты (например, буторфанол — антагонист μ и агонист κ опиоидных рецепторов).
Другие «мишени» для лекарственных веществ
К другим «мишеням» относят ионные каналы, ферменты, транспортные белки.
Ионные каналы. Одной из основных «мишеней» для ЛВ являются потенциалзависимые ионные каналы, избирательно проводящие Na + , Са 2+ , К + и другие ионы через клеточную мембрану. В отличие от рецептор-управляемых ионных каналов, открываемых при взаимодействии вещества с рецептором, эти каналы регулируются потенциалом действия (открываются при деполяризации клеточной мембраны). ЛВ могут или блокировать потенциалзависимые ионные каналы и таким образом нарушать поступление через них ионов, или активи- ровать, т.е. способствовать прохождению ионных токов. Большинство ЛВ блокируют ионные каналы.
Местные анестетики блокируют потенциалзависимые Nа + -каналы. К числу блокаторов Na + -каналов относятся и многие противоаритмические средства (хинидин, лидокаин, прокаинамид). Некоторые противоэпилептические средства (фенитоин, карбамазепин) также блокируют потенциалзависимые Nа + -каналы, и с этим связана их противосудорожная активность. Блокаторы натриевых каналов нарушают вхождение в клетку Na + и таким образом препятствуют деполяризации клеточной мембраны.
Весьма эффективными при лечении многих сердечно-сосудистых заболеваний (гипертонической болезни, сердечных аритмий, стенокардии) оказались блокаторы Са 2+ -каналов (нифедипин, верапамил и др.). Ионы кальция принимают участие во многих физиологических процессах: в сокращении гладких мышц, генерации импульсов в синусно-пред- сердном узле и проведении возбуждения по предсердно-желудочковому узлу, агрегации тромбоцитов и др. Блокаторы медленных кальциевых
каналов препятствуют вхождению ионов кальция внутрь клетки через потенциалзависимые каналы и вызывают расслабление гладких мышц сосудов, уменьшение частоты сокращений сердца и АВ-проводимости, нарушают агрегацию тромбоцитов. Некоторые блокаторы кальциевых каналов (нимодипин, циннаризин) преимущественно расширяют сосуды мозга и оказывают нейропротекторное действие (препятствуют поступлению избыточного количества Са 2+ внутрь нейронов).
В качестве лекарственных средств используются как активаторы, так и блокаторы калиевых каналов. Активаторы калиевых каналов (миноксидил) нашли применение в качестве антигипертензивных средств. Они способствуют выходу ионов калия из клетки, что приводит к гиперполяризации клеточной мембраны и уменьшению тонуса гладких мышц сосудов. В результате происходит снижение артериального давления. ЛВ, блокирующие потенциалзависимые калиевые каналы (амиодарон, соталол), нашли прменение при лечении аритмий сердца. Они препятствуют выходу К + из кардиомиоцитов, вследствие чего увеличивают продолжительность потенциала действия и удлиняют эффективный рефрактерный период (ЭРП). Блокада АТФ-зависимых калиевых каналов в β-клетках поджелудочной железы приводит к повышению секреции инсулина; блокаторы этих каналов (производные сульфонилмочевины) применяют как противодиабетические средства.
Ферменты. Многие ЛВ являются ингибиторами ферментов. Ингибиторы МАО нарушают метаболизм (окислительное дезаминирование) катехоламинов (норадреналина, дофамина, серотонина) и повышают их содержание в ЦНС. На этом принципе основано действие антидепрессантов — ингибиторов МАО (например, ниаламида). Механизм действия нестероидных противовоспалительных средств связан с ингибированием циклооксигеназы, в результате снижается биосинтез простагландинов Е 2 и I 2 и развивается прововоспалитель- ное действие. Ингибиторы ацетилхолинэстеразы (антихолинэстеразные средства) препятствуют гидролизу ацетилхолина и повышают его содержание в синаптической щели. Препараты этой группы применяют для повышения тонуса гладкомышечных органов (ЖКТ, мочевого пузыря) и скелетных мышц.
Транспортные системы. ЛВ могут действовать на транспортные системы (транспортные белки), переносящие молекулы некоторых веществ или ионы через мембраны клеток. Например, трициклические антидепрессанты блокируют транспортные белки, которые переносят норадреналин и серотонин через пресинаптическую мемб-
рану нервного окончания (блокируют обратный нейрональный захват норадреналина и серотонина). Сердечные гликозиды блокируют К + -АТФазу мембран кардиомиоцитов, осуществляющую транспорт Na + из клетки в обмен на К + .
Возможны и другие «мишени», на которые могут действовать ЛС. Так, антацидные средства нейтрализуют соляную кислоту желудка, их применяют при повышенной кислотности желудочного сока (гиперацидном гастрите, язвенной болезни желудка).
Перспективной «мишенью» для ЛС являются гены. С помощью избирательно действующих ЛС возможно оказывать прямое влияние на функцию определенных генов.
2.2. ВИДЫ ДЕЙСТВИЯ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ
Различают следующие виды действия: местное и резорбтивное, рефлекторное, прямое и косвенное, основное и побочное и некоторые другие.
Местное действие ЛВ оказывает при контакте с тканями в месте его нанесения (обычно это кожа или слизистые оболочки). Например, при поверхностной анестезии местный анестетик действует на окончания чувствительных нервов только в месте нанесения на слизистую оболочку. Для оказания местного действия ЛВ назначают в форме мазей, примочек, полосканий, пластырей. При назначении некоторых ЛВ в виде глазных или ушных капель также рассчитывают на их местное действие. Однако какое-то количество ЛВ обычно всасывается с места нанесения в кровь и оказывает общее (резорбтивное) действие. При местном применении ЛВ возможно также рефлекторное действие.
Резорбтивное действие (от лат. resorbeo — поглощаю) — эффекты, вызываемые ЛВ после всасывания в кровь или непосредствен- ного введения в кровеносный сосуд и распределения в организме. При резорбтивном действии, как и при местном, вещество может возбуждать чувствительные рецепторы и вызывать рефлекторные реакции.
Рефлекторное действие. Некоторые ЛВ способны возбуждать окончания чувствительных нервов кожи, слизистых оболочек (экстерорецепторы), хеморецепторы сосудов (интерорецепторы) и вызывать рефлекторные реакции со стороны органов, расположенных в удалении от места непосредственного контакта вещества с чувствительными рецепторами. Примером возбуждения экстерорецепторов
кожи эфирным горчичным маслом является действие горчичников. Лобелин при внутривенном введении возбуждает хеморецепторы сосудов, что приводит к рефлекторной стимуляции дыхательного и сосудодвигательного центров.
Прямое (первичное) действие ЛВ на сердце, сосуды, кишечник и другие органы развивается при непосредственном воздействии на эти органы. Например, сердечные гликозиды вызывают кардиотонический эффект (усиление сокращений миокарда) вследствие их непосредственного влияния на кардиомиоциты. Вызываемое же сердечными гликозидами повышение диуреза у больных с сердечной недостаточностью обусловлено увеличением сердечного выброса и улучшением гемодинамики. Такое действие, при котором ЛВ изменяет функцию одних органов, воздействуя на другие органы, обозначают как косвенное (вторичное) действие.
Основное действие. Действие, ради которого применяют ЛВ при лечении данного заболевания. Например, фенитоин обладает противосудорожными и антиаритмическими свойствами. У больного эпилепсией основное действие фенитоина — противосудорожное, а у больного с сердечной аритмией, вызванной передозировкой сердечных гликозидов, — антиаритмическое.
Все остальные (кроме основного) эффекты ЛВ, возникающие при его приеме в терапевтических дозах, расценивают как п о б о ч н о е действие. Эти эффекты часто бывают неблагоприятными (отрицательными) (см. главу «Побочное и токсическое действие лекарственных веществ»). Например, ацетилсалициловая кислота может вызвать изъязвление слизистой оболочки желудка, антибиотики из группы аминогликозидов (канамицин, гентамицин и др.) — нарушение слуха. Отрицательное побочное действие часто служит причиной ограничения применения того или иного ЛВ и даже исключения его из списка лекарственных препаратов.
Избирательное действие ЛВ направлено преимущественно на один орган или систему организма. Так, сердечные гликози- ды обладают избирательным действием на миокард, окситоцин — на матку, снотворные средства — на ЦНС.
Центральное действие развивается вследствие прямого влияния ЛВ на ЦНС. Центральное действие характерно для веществ, проникающих через ГЭБ. Для снотворных средств, антидепрессантов, анксиолитиков, средств для наркоза это основное действие. В то же время центральное действие может быть побочным (нежелательным).
Так, многие антигистаминные средства вследствие центрального действия вызывают сонливость.
Периферическое действие обусловлено влиянием ЛВ на периферический отдел нервной системы или на органы и ткани. Курареподобные средства (миорелаксанты периферического действия) расслабляют скелетные мышцы, блокируя передачу возбуждения в нервно-мышечных синапсах, некоторые периферические вазодилататоры расширяют кровеносные сосуды, действуя непосредственно на гладкомышечные клетки. Для веществ с основным центральным действием периферические эффекты обычно побочные. Например, антипсихотическое средство хлорпромазин вызывает расширение сосудов и снижение АД (нежелательное действие), блокируя периферические α-адренорецепторы.
Обратимое действие является следствием обратимого связывания ЛВ с «мишенями» (рецепторами, ферментами). Действие такого вещества можно прекратить путем его вытеснения из связи c «мишенью» другим ЛВ.
Необратимое действие возникает, как правило, в результате прочного (ковалентного) связывания ЛВ с «мишенями». Например, ацетилсалициловая кислота необратимо блокирует циклооксигеназу, поэтому действие препарата прекращается лишь после синтеза нового фермента.
агонистов и антагонистов рецепторов | BioAspect
Рецепторы
Рецепторы— это трансмембранные белки, которые обеспечивают передачу химических сигналов из внешнего микроокружения клетки в клетки, а также внутри клетки. Рецепторы обычно встроены в мембрану и обращены либо вне клеток (внеклеточные рецепторы), либо в цитозоль (цитозольные рецепторы). Они также могут находиться в ядре клетки (ядерные рецепторы или факторы транскрипции).
Интересный факт: у нематоды C. elegans имеется 270 ядерных рецепторов. H umans, мыши и крысы, однако, имеют соответственно 48, 49 и 47 ядерных рецепторов каждый !
Рецепторыпозволяют осуществлять межклеточную коммуникацию, а также регулировать многие клеточные биохимические процессы (транскрипцию генов, фосфорилирование белков, активацию ферментов и т. Д.). Состояние активации рецептора изменяется, когда рецептор связывается с лигандом (т.е.эндогенные лиганды могут быть пептидами, гормонами, нейротрансмиттерами, витаминами A и D или другими соединениями). Связывание лиганда обычно запускает конформационное изменение рецептора, которое может активировать или деактивировать рецептор и инициировать каскад событий, необходимых для получения биологического функционального эффекта. В некоторых случаях занятый рецептор фактически сам выражает биологическую активность. Например, занятый рецептор может приобретать ферментативную активность или становиться активным ионным каналом.
Ксенобиотические лиганды, которые вызывают те же биологические эффекты, что и природные лиганды, называются «агонистами рецепторов» . И наоборот, «антагонисты рецептора» — это лиганды, которые ингибируют биологическую активность рецептора. Обратите внимание, что концепция агонизма и антагонизма рецепторов относится только к взаимодействию между рецепторами и лигандами, а не к их биологическим эффектам.
См. Наш каталог агонистов и антагонистов рецепторов
Агонисты рецепторов
Агонист является миметиком природного лиганда и при связывании с рецептором оказывает такое же биологическое действие, как и природный лиганд.Агонисты связываются с рецептором в том же сайте связывания, что и природный лиганд, и приводит к полной (обычные агонисты) или частичной (частичные агонисты) активации . Обычные агонисты увеличивают долю активированных рецепторов. Обратные агонисты представляют собой агенты, которые связываются с тем же рецептором, что и агонист, но вызывают фармакологический ответ, противоположный этому агонисту. Термин «обратный агонистический» ответ имеет смысл только в том случае, если несвязанный свободный рецептор имеет базальную или конститутивную активность.
Обычный агонист увеличивает активность рецептора выше его базального уровня, тогда как обратный агонист снижает активность ниже базального уровня. Примером рецептора, который обладает базовой активностью и для которого были идентифицированы обратные агонисты, является рецептор ГАМК. Агонисты рецептора ГАМК (например, бензодиазепины) создают седативный эффект, тогда как обратные агонисты (например, Ro15-4513) обладают анксиогенным действием или даже судорожным действием (некоторые бета-карболины).
Нейтральный антагонист не проявляет активности в отсутствие агониста или обратного агониста, но может блокировать активность любого из них. Эффективность полного агониста по определению составляет 100%, нейтральный антагонист имеет эффективность 0%, а обратный агонист имеет эффективность <0% (т. Е. Отрицательную) (см. График ниже).
Щелкните здесь, чтобы увидеть рецептор Агонисты от BioAspect и наших высококачественных поставщиков
Антагонисты рецепторов
Антагонисты рецептораявляются ингибиторами рецепторной активности.Антагонисты имитируют лиганды, которые связываются с рецептором и предотвращают активацию рецептора естественным лигандом. Предотвращение активации может иметь множество последствий. Если связывание природного агониста с рецептором приводит к усилению клеточной функции, антагонист, который связывает и блокирует этот рецептор, снижает функцию. Антагонисты усиливают клеточную функцию, если они блокируют действие вещества, которое обычно снижает клеточную функцию. И наоборот, антагонисты снижают клеточную функцию, если они блокируют действие вещества, которое обычно увеличивает клеточную функцию.Антагонисты рецепторов могут быть классифицированы как обратимые или необратимые . Обратимые антагонисты легко диссоциируют от своего рецептора; необратимые антагонисты образуют стабильную химическую связь со своим рецептором (например, путем алкилирования). Псевдо-необратимые антагонисты со временем медленно диссоциируют от своего рецептора. Описаны три типа антагонистов рецептора: конкурентный (связывание с активным сайтом рецептора), неконкурентный (связывание с сайтом, отличным от активного сайта или аллостерическим сайтом рецептора) и неконкурентный .Неконкурентные антагонисты отличаются от неконкурентных антагонистов тем, что они требуют активации рецептора агонистом, прежде чем они смогут связываться с отдельным аллостерическим сайтом связывания.
Нажмите здесь, чтобы увидеть рецепторы Антагонисты от BioAspect и наших высококачественных поставщиков
Агонисты и антагонисты — UTS Pharmacology
Злодей, который помогает нашему телу? Агонисты и антагонисты (черновой вариант)
Когда мы говорим об антагонистах и агонистах, нет, мы не говорим о злодеях из кино, пытающихся уничтожить половину населения Вселенной, вместо этого мы говорим о наркотиках, которые взаимодействуют с рецепторами нашего тела.
агонисты и антагонисты; обсудить ключевые различия между ними.
Агонист — это лекарство, которое связывается с рецептором, вызывая аналогичный ответ на предполагаемое химическое вещество и рецептор. В то время как антагонист — это лекарство, которое связывается с рецептором либо на первичном сайте, либо на другом сайте, что все вместе останавливает рецептор от выработки ответа.
Основное различие между этими двумя препаратами состоит в том, что один имитирует предполагаемую реакцию, когда антагонист связывается с рецептором и останавливает / замедляет ответы.Агонисты по существу имитируют активность нормальных нейромедиаторов, таких как ацетилхолин, и имитируют аналогичный ответ рецепторов, с которыми они связываются. Прекрасная аналогия — это торговый автомат. Обычно, чтобы купить напиток, вы вставляете монету в 1 доллар в автомат, и в ответ он выплевывает вашу любимую газировку. Агонистом в этом сценарии было бы использование металлического диска того же размера, что и монета, для вставки в автомат, таким образом, используя ту же прорезь для монет с имитацией монеты, чтобы получить газировку.
Антагонист действует противоположно агонисту. Он связывается с рецепторами и мешает рецептору производить желаемый ответ. Возвращаясь к аналогии, это похоже на заклинивание гнезда для монет в автомате, так что он не может выполнять свою функцию, пока блокировка не будет устранена.
–Выберите антагонист, обсудите первичный механизм ингибирования и подробно объясните, как это связано со способом, которым он взаимодействует с рецепторами. — [ краткое описание предмета, которое я могу изменить, если смогу найти более интересный антагонист]
Атропин — это обратимый конкурентный антагонист мускариновых рецепторов ацетилхолина, на языке, который мы с вами понимаем, это съемный блокиратор монет для торгового автомата, который контролирует некоторые из наших функций организма, такие как слюноотделение и частоту сердечных сокращений. [изображение модели замка и ключа]
Атропин подавляет некоторые функции парасимпатической системы, поэтому он влияет на такие вещи, как частота сердечных сокращений, слюноотделение и расширение зрачков. Атропин используется для лечения брадикардии, то есть замедления сердечного ритма, у пациентов, а также для снижения количества слюны, вырабатываемой при некоторых конкретных операциях.
(Посещали 3196 раз, сегодня 57 посещений)
опиоидных агонистов, частичных агонистов, антагонистов: Oh My!
Когда дело доходит до прогнозирования терапевтических или побочных эффектов (и эффективности), специфическая фармакодинамика опиоидов играет важную роль.Как мы все хорошо знаем, фармакодинамика — это изучение того, что лекарство делает с организмом, включая участие в связывании рецепторов и конформационных изменениях, которые в конечном итоге приводят к терапевтическим и нетерапевтическим эффектам (т.е. побочным эффектам). Некоторые из побочных эффектов опиоидов перечислены в таблице 1 . 1Таблица 1. Побочные эффекты, связанные с употреблением опиоидов 1
| Общие побочные эффекты | Тяжелые побочные эффекты |
| Когнитивные нарушения Запор Пониженный тестостерон / сексуальная дисфункция Тошнота / рвота Ортостатическая гипотензия Седативный эффект / сонливость | Наркомания Угнетение дыхания, вызванное опиоидами |
Выявлено 4 типа опиоидных рецепторов: мю, дельта, каппа и опиоидный рецептор, подобный-1 (ORL-1).Некоторые рецепторы можно разделить на подтипы. Эти рецепторы важны для выражения передачи боли и модуляции путей, включая нейротрансмиссию в лимбической системе, среднем мозге, спинном мозге и таламусе. Опиоидные рецепторы повсеместно присутствуют по всему телу, включая, помимо прочего, желудочно-кишечный тракт (ЖКТ), иммунные клетки, гипофиз и кожу. На этих участках опиоидные рецепторы выполняют различные обезболивающие и неанальгетические функции. Сводка фармакологических и физиологических эффектов для каждого опиоидного рецептора приведена в таблице 2 . 2-4
Таблица. 2. Обобщенные эффекты отдельных опиоидных рецепторов 2-4
| му | Дельта | Каппа | ОРЛ-1 | |
| Клинические эффекты | Анальгезия Депрессия Эйфория Физическая зависимость Респираторный Седация | Анальгезия Подавляет высвобождение дофамина Модуляция рецептора мю | Анальгезия Диурез Дисфория | Анальгезия Седация |
Большинство опиоидов являются агонистами мю с различной активностью в отношении каппа-рецепторов.
Опиоидные рецепторы — это 7 трансмембранных белков, которые связаны с ингибирующими G-белками. При активации они уменьшают выработку аденилциклазы вторичного мессенджера циклического аденозинмонофосфата. Это вызывает уменьшение притока кальция из-за ингибирования потенциал-управляемых кальциевых каналов и приводит к активации калиевых каналов, что приводит к гиперполяризации. Гиперполяризованное состояние вызывает подавление нейрональной передачи сигналов, что в данном случае подавляет передачу боли. 2,5
Опиоиды классифицируются по категориям в зависимости от связывания с рецептором и сродства (, таблица 3, ). Эти классификации являются агонистами, частичными агонистами и антагонистами. Есть опиоиды, которые обладают двойными агонистическими и антагонистическими функциями.
Таблица 3. Примеры опиоидов по связыванию рецепторов
| Полный агонист | Частичный агонист | Смешанный агонист | Антагонист |
| Кодеин Фентанил Героин Гидрокодон Гидроморфон Леворфанол Меперидин Метадон Морфин Оксикодон Оксиморфон | Бупренорфин Буторфанол Пентазоцин Трамадол | Бупренорфин Буторфанол Нальбуфин Пентазоцин | Налоксон Налтрексон |
Полные агонисты прочно связываются с опиоидными рецепторами и претерпевают значительные конформационные изменения для достижения максимального эффекта.Примеры полных агонистов включают кодеин, фентанил, героин, гидрокодон, метадон, морфин и оксикодон.
Частичные агонисты вызывают меньшие конформационные изменения и активацию рецепторов, чем полные агонисты. В низких дозах как полные, так и частичные агонисты могут оказывать аналогичные эффекты со своими собратьями-полными агонистами. Однако, когда доза частичных агонистов увеличивается, анальгетическая активность выйдет на плато, и дальнейшее увеличение доз не принесет дополнительного облегчения, но может усилить побочные эффекты.Примеры частичных агонистов включают бупренорфин, буторфанол и трамадол.
Существуют смешанные агонисты / антагонисты, которые демонстрируют разную активность в зависимости от опиоидного рецептора, но также разную дозу. Примеры включают бупренорфин, буторфанол, налбуфин и пентазоцин. И некоторые опиоиды являются агонистами одного или нескольких опиоидных рецепторов, но также антагонистами других опиоидных рецепторов. Обобщение рецепторных эффектов агонистов / антагонистов можно найти в Таблице 4. 4
Таблица 4. Рецепторный эффект смешанных опиоидных агонистов / антагонистов 4
| Опиоиды | му | Каппа | Дельта |
| Пентазоцин | Частичный агонист | Агонист | |
| Нальбуфин | Антагонист | Агонист | |
| Бупренорфин | Частичный агонист | Антагонист | Антагонист (слабый) |
| Буторфанол | Антагонист | Частичный агонист |
Пентазоцин одобрен FDA и показан для обезболивания и содержит ацетаминофен или налоксон.Механизм действия — частичный агонист мю-опиоидного рецептора и полный агонист каппа-опиоидного рецептора. Хотя пентазоцин слабо противодействует анальгетическим эффектам полных агонистов, он также вызывает неполное обращение депрессии поведения, сердечно-сосудистой системы и дыхания, вызванной морфином и другими полными агонистами. 6
Нальбуфин представляет собой синтетический анальгетический опиоид, проявляющий агонистическую активность в отношении каппа-рецептора, одновременно действуя как антагонист в отношении мю-рецептора.Налбуфин оказывает максимальное воздействие на угнетение дыхания в дозах более 30 мг. Сообщалось, что налбуфин обращает вспять угнетение дыхания, но не анальгезирует мю-агонисты. 7,8 Нальбуфин может вызвать синдром отмены у пациентов, которые физически зависят от агонистов мю-опиоидов. 9
Бупренорфин показан в высоких дозах при расстройствах, связанных с употреблением опиоидов, а в более низких дозах — для лечения умеренной и сильной боли. Препараты бупренорфина, одобренные Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) для лечения боли, включают белбука, бупренекс и бутранс. 10-12 Препараты бупренорфина, одобренные для лечения опиоидной зависимости, включают бунавайл, пробуфин, субоксон, субутекс и зубсолв. 13-17 Бупренорфин в 25–100 раз сильнее морфина. 18 Бупренорфин проявляет частичное агонистическое поведение в отношении мю-рецептора и проявляет антагонистическое поведение в отношении каппа-рецептора. Бупренорфин имеет сильное сродство с мю-рецептором, вызывая сильное связывание и, следовательно, конкуренцию с рецептором, вытесняя другие опиоиды, такие как метадон и морфин.Кроме того, происходит неполная диссоциация мю-рецептора, вызывающая длительную активность рецептора. 18 Сродство количественно определяют с использованием значений Ki, и чем меньше значение Ki, тем сильнее сродство связывания с рецептором. Аффинность связывания мю бупренорфина по сравнению с другими опиоидами можно найти в Таблице 5. Следует отметить, что бупренорфин имеет более высокое сродство связывания по сравнению с налоксоном и, следовательно, в более высоких дозах, когда наиболее вероятно злоупотребление бупренорфином, а не налоксоном. .Только при более низких дозах наблюдается некоторое конкурентное связывание, и только в этом случае мы должны разумно ожидать некоторого улучшения с помощью высоких доз налоксона непрерывной инфузии. Это, конечно, вызывает вопрос о полезности комбинированного продукта, субоксона.
Таблица 5. Сродство различных опиоидов к мю-рецепторам 19
| Опиоиды | Диапазон значений Ki |
| Леворфанол | 0.19 к .23 32 |
| Бупренорфин | от 0,21 до 1,5 |
| Налтрексон | от 0,4 до 0,6 (антагонистические эффекты) 20 |
| Фентанил | от 0,7 до 1,9 |
| Метадон | от 0,72 до 5,6 |
| Налоксон | от 1 до 3 (эффекты антагониста) 20 |
| Морфин | 1.02 по 4 |
| Пентазоцин | от 3,9 до 6,9 |
| Кодеин | 65 по 135 |
Из-за частичного агонизма увеличение доз влияет на плато угнетения дыхания, что делает его жизнеспособным вариантом для людей с повышенным риском угнетения дыхания. 21 Антагонизм каппа-рецепторов имеет преимущества для снижения вызванного стрессом поведения, связанного с поиском лекарств, из-за последующей блокады динорфинов.Динорфины — это опиоидные пептиды, селективные к каппа-рецепторам, которые вызывают беспокойство, стресс и увеличивают желание употреблять опиоиды. Кроме того, антагонизм каппа продемонстрировал антидепрессивные свойства. 22,23
Буторфанол является антагонистом мю-опиоидов с низкой внутренней активностью и агонистом каппа-опиоидов, проявляющим высокое сродство. Буторфанол показан для снятия боли у пациентов, у которых альтернативные варианты лечения неэффективны, непереносимы или неадекватны, и представлен в виде назального спрея и инъекций.Из-за высокого сродства к каппа-рецепторам он может вызывать психотомиметические эффекты. При более высоких дозах степень угнетения дыхания существенно не увеличивается, как у большинства смешанных агонистов / антагонистов. Однако продолжительность угнетения дыхания больше из-за преобладающих физиологических реакций, опосредованных мю, по сравнению с каппа. 23-25
Агонисты опиоидов также связываются с периферическими мю-опиоидными рецепторами в желудочно-кишечном тракте, вызывая нарушение моторики и секреции, что приводит к запорам.Антагонисты мю-опиоидных рецепторов периферического действия (PAMORA) действуют конкретно в желудочно-кишечном тракте для борьбы с запорами. Однако периферические антагонисты мю-опиоидов не проникают через гематоэнцефалический барьер, что позволяет избежать блокады центрально-опосредованной анальгезии и других центрально-опосредованных эффектов агонистов опиоидов. Примеры ПАМОР включают алвимопан (Энтерег), метилналтрексон (Релистор) и налоксегол (Мовантик). Последние 2 рекомендованы FDA при запорах, вызванных опиоидами, обычно после того, как безрецептурные слабительные не действуют.Однако Алвимопан показан для периоперационного лечения послеоперационной кишечной непроходимости, чтобы ускорить восстановление верхних и нижних отделов ЖКТ после операции. Кроме того, в отличие от всех других ПАМОР, у Алвимопана есть предупреждение о потенциальном риске инфаркта миокарда при длительном применении, что не является признанным риском для других ПАМОР. 26-28
Наконец, как и в любой великой истории, есть антагонисты. Однако в истории с опиоидами антагонисты опиоидов могут спасти жизни.Налтрексон является антагонистом опиоидов, который блокирует действие опиоидов путем конкурентного связывания. Налтрексон показан при алкогольной и опиоидной зависимости и полезен, потому что его блокада опиоидных рецепторов вторично снижает активность дофамина, которая в противном случае усиливается алкоголем. 29 Налоксон является антагонистом мю-опиоидных рецепторов и реверсивным агентом, который используется для снижения риска индуцированного опиоидами угнетения дыхания путем вытеснения полных опиоидных агонистов. Доступные составы — это наркан (назальный), Evzio (автоинъектор) и раствор для инъекций, последний из которых часто вводят интраназально без указания инструкции, прикрепив распылитель к концу шприца. 30,31 Членам семьи, друзьям и случайным прохожим теперь разрешено осуществлять административную деятельность в большинстве штатов (защищенных законами Доброго Самаритянина) в дополнение к профессиональным специалистам по оказанию первой помощи, например парамедикам.
Хотя химический состав опиоидов описан, существуют и другие параметры, которые также необходимо учитывать для определения индивидуальной реакции. К ним относятся возраст, сопутствующие заболевания, тяжесть заболевания, пол, генетика и вес, все из которых могут положительно или отрицательно повлиять на реакцию на лекарство.В дополнение к описанной здесь фармакодинамике многие синтетические опиоиды обладают дополнительными механизмами действия, такими как блокада норадренергического обратного захвата и ингибирование рецепторов n-метил-D-аспартазы (NMDA). По этой причине, как это ошибочно подразумевается в недавних рекомендациях CDC, неудача и / или непереносимость одного опиоида не обязательно приравнивается к неудаче другого. Поэтому важно учитывать все эти переменные при принятии решений, влияющих на выбор или прекращение приема опиоидов.
Эта статья является исключительной работой авторов, и высказанные мнения / утверждения не отражают мнение перечисленных работодателей, аффилированных лиц сотрудников и / или фармацевтических компаний.
Ребекка Чу — кандидат фармацевтических наук, специализируется в области экономики и здравоохранения в Колледже фармации и здравоохранения Олбани (Нью-Йорк).
Александра Чиани — кандидат фармацевтических наук в Фармацевтическом колледже Университета Западной Новой Англии в Спрингфилде, Массачусетс. Она работает стажером в аптеке Hannaford Supermarket Pharmacy в Роттердаме, Нью-Йорк.
Mena Raouf, PharmD, проживает в медицинском центре Stratton VA в Олбани, штат Нью-Йорк.Он закончил резидентуру PGY-1 в системе здравоохранения VA Tennessee Valley в Нэшвилле и получил степень PharmD в Колледже фармации и медицинских наук Олбани (Нью-Йорк).
Список литературы
- Беньямин Р., Трескот А.М., Датта С. и др. Опиоидные осложнения и побочные эффекты. Врач-болеутоляющий. 2008; 11 (2 доп.): S105-20.
- Вальехо Р., Баркин Р.Л., Ван В.К. Фармакология опиоидов в лечении хронических болевых синдромов. Врач-болеутоляющий. 2011; 14 (4): E343-60.
- Waldhoer M, Bartlett SE, Whistler JL. Опиоидные рецепторы. Annu Rev Biochem. 2004; 73: 953–90.
- Нельсон Л.С., Олсен Д. Опиоиды. В: Hoffman RS, Howland M, Lewin NA, Nelson LS, Goldfrank LR. ред. Экстренная токсикологическая помощь Голдфрэнка. 10-е изд. Нью-Йорк, штат Нью-Йорк: Макгроу-Хилл; 2015.
- Аль-Хасани Р., Брухас MR. Молекулярные механизмы передачи сигналов и поведения, зависимых от опиоидных рецепторов. Анестезиология. 2011; 115 (6): 1363-81. DOI: 10.1097 / ALN.0b013e318238bba6.
- Talwin [информация о назначении]. Бриджуотер, Нью-Джерси: Санофи-авентис; 2011. accessdata.fda.gov/drugsatfda_docs/label/2011/018733s015lbl.pdf. По состоянию на 5 января 2018 г.
- Freye E, Azevedo L, Hartung E. Устранение угнетения дыхания, связанного с фентанилом, с помощью налбуфина. Влияние на кривую реакции на CO2 у человека. Acta Anaesthesiol Belg. 1985; 36 (4): 365-74.
- Blaise GA, Nugent M, McMichan JC Durant, PA.Побочные эффекты налбуфина при обращении вызванного опиоидами угнетения дыхания: сообщение о четырех случаях. Can J Anaesth. 1990; 37 (7): 794-7
- Нубаин [информация о назначении]. Честнат-Ридж, штат Нью-Йорк: Par Pharmaceutical; 2016. accessdata.fda.gov/drugsatfda_docs/label/2016/018024s041lbl.pdf. По состоянию на 5 января 2018 г.
- Buprenex [информация о назначении]. Норт-Честерфилд, штат Вирджиния: Indivior; 2017. accessdata.fda.gov/drugsatfda_docs/label/2017/209819s000lbl.pdf.Доступ 5 января 2018 г.
- Бутранс [информация о назначении]. Стэмфорд, Коннектикут: Purdue Pharma; 2014. accessdata.fda.gov/drugsatfda_docs/label/2014/021306s015s019lbl.pdf. По состоянию на 5 января 2018 г.
- Belbuca [информация о назначении]. Малверн, Пенсильвания: Endo Pharmaceuticals; 2015. accessdata.fda.gov/drugsatfda_docs/label/2015/207932s000lbl.pdf. По состоянию на 5 января 2018 г.
- Субутекс [информация о назначении]. Колумбус, Огайо: Roxane Laboratories; 2015. док.boehringer-ingelheim.com/Prescribing%20Information/PIs/Roxane/Buprenorphine%20HCl%20Sublingual%20Tabs/10004964_01%20Buprenorphine%20HCl%20Sublingual%20Tabs.pdf. По состоянию на 5 января 2018 г.
- Bunavail [информация о назначении]. Роли, Северная Каролина: BioDelivery Sciences International, Inc; 2014. accessdata.fda.gov/drugsatfda_docs/label/2014/205637s000lbl.pdf. По состоянию на 5 января 2018 г.
- Субоксон [информация о назначении]. Ричмонд, Вирджиния: Reckitt Benckiser Pharmaceuticals; 2010 г.accessdata.fda.gov/drugsatfda_docs/label/2010/022410s000lbl.pdf. По состоянию на 5 января 2018 г.
- Zubsolv [инструкция по применению]. Морристаун, Нью-Джерси: Orexo US, Inc; 2017. zubsolv.com/wp-content/uploads/2015/01/ZubsolvFullPrescribingInformation.pdf. По состоянию на 5 января 2018 г.
- Пробуфин [информация о назначении]. Принстон (Нью-Джерси): Braeburn Pharms, Inc; 2016. accessdata.fda.gov/drugsatfda_docs/label/2016/204442Orig1s000lbl.pdf. По состоянию на 5 января 2018 г.
- Лютфи К., Коуэн А.Бупренорфин: уникальный препарат со сложной фармакологией. Curr Neuropharmacol. 2004; 2 (4): 395-402. DOI: 10.2174 / 15701559477.
- PDSP Ki Database pdsp.unc.edu/databases/pdsp.php?recDDRadio=recDDRadio&receptorDD=1&receptor=&speciesDD=&speciesFreeRadio=speciesFreeRadio&species=human&sourceDDRadio=sourceDDRadio&sourcesDD=&source=&hotDDRadio=hotDDRadio&hotLigandDD=&hotLigand=&testDDRadio=testDDRadio&testLigandDD=&testLigand=&refDDRadio=refDDRadio&referenceDD= & reference = & KiGreater = & KiLess = & kiAllRadio = все & doQuery = Отправить + Запрос.По состоянию на 5 января 2018 г.
- Wang D, Sun X, Sadee W. Различные эффекты опиоидных антагонистов на μ-, δ- и κ-опиоидные рецепторы с предварительной обработкой агонистами и без нее. J Pharmacol Exp Ther. 2007; 321 (2): 544-52.
- Дахан А. Респираторные эффекты, вызванные опиоидами: новые данные о бупренорфине. Palliat Med. 2006; 20 Приложение 1: s3-8.
- Маклафлин Дж. П., Мартон-Поповичи М, Чавкин С (2003). Антагонизм каппа-опиоидных рецепторов и нарушение гена продинорфина блокируют вызванные стрессом поведенческие реакции. J Neurosci. 23 (13): 5674–83.
- Психическое здоровье ежедневно. 2 новых антагониста каппа-опиоидных рецепторов (KOR) при депрессии. mentalhealthdaily.com/2014/12/19/2-new-kappa-opioid-receptor-kor-antagonists-for-depression/. По состоянию на 5 января 2018 г.
- Stadol [информация о назначении]. Дейтон, Нью-Джерси: Женева Фармасьютикалз, Инк; 2002. accessdata.fda.gov/drugsatfda_docs/label/2002/19890s17lbl.pdf. По состоянию на 5 января 2018 г.
- Walsh SL, Chausmer AE, Strain EC, Bigelow GE.Оценка мю- и каппа-опиоидного действия буторфанола у людей посредством дифференциальной блокады налтрексона. Психофармакология (Берл). 196 (1): 143-55
- Мовантик [инструкция по применению]. Уилмингтон, Делавэр: AstraZeneca Pharmaceuticals, LP; 2017. azpicentral.com/movantik/movantik.pdf. По состоянию на 5 января 2018 г.
- Релистор [предписывающая информация]. Бриджуотер, штат Нью-Джерси: Salix Pharmaceuticals; 2017. shared.salix.com/shared/pi/relistor-pi.pdf. По состоянию на 5 января 2018 г.
- Entereg [информация о назначении]. Станция Уайтхаус, Нью-Джерси: Merck & Co; 2015. merck.com/product/usa/pi_circulars/e/entereg/entereg_pi.pdf. По состоянию на 5 января 2018 г.
- Revia [информация о назначении]. Помона, штат Нью-Йорк: Barr Pharmaceuticals, Inc; 2013. accessdata.fda.gov/drugsatfda_docs/label/2013/018932s017lbl.pdf. По состоянию на 5 января 2018 г.
- Наркан [информация о назначении]. Рэднор, Пенсильвания: Adapt Pharma, Inc; 2015. accessdata.fda.gov/drugsatfda_docs/label/2015/208411lbl.pdf. По состоянию на 5 января 2018 г.
- Evzio [информация о назначении]. Ричмонд, Вирджиния: Калео; 2014. accessdata.fda.gov/drugsatfda_docs/label/2014/205787Orig1s000lbl.pdf. По состоянию на 5 января 2018 г.
- Prommer E. Леворфанол: возвращение к недостаточно используемому анальгетику. Паллиативная помощь: исследования и лечение . 2014 Янв; 8: PCRT-S13489.
Объяснитель: как действуют наркотики?
Независимо от того, выписано ли лекарство врачом, куплено без рецепта или получено незаконным путем, мы обычно воспринимаем их механизм действия как должное и верим, что они сделают то, что должны.
Но как таблетки ибупрофена снимают головную боль? И что делает антидепрессант, чтобы помочь сбалансировать химию вашего мозга?
Для того, что кажется таким невероятным, механика наркотиков удивительно проста. В основном речь идет о рецепторах и молекулах, которые их активируют.
Рецепторы
Рецепторы— это большие белковые молекулы, встроенные в клеточную стенку или мембрану. Они получают (отсюда «рецепторы») химическую информацию от других молекул, таких как лекарства, гормоны или нейротрансмиттеры, вне клетки.
Эти внешние молекулы связываются с рецепторами клетки, активируя рецептор и генерируя биохимический или электрический сигнал внутри клетки. Затем этот сигнал заставляет клетку делать определенные вещи, например заставлять нас чувствовать боль.
Агонистические препараты
Те молекулы, которые связываются со специфическими рецепторами и заставляют процесс в клетке становиться более активным, называются агонистами. Агонист — это то, что вызывает в клетке определенный физиологический ответ. Они могут быть натуральными или искусственными.
Например, эндорфины являются естественными агонистами опиоидных рецепторов. Но морфин — или героин, который превращается в морфин в организме — является искусственным агонистом основного опиоидного рецептора.
Искусственный агонист настолько структурно похож на естественный агонист рецептора, что может оказывать такое же действие на рецептор. Многие лекарства созданы для имитации природных агонистов, чтобы они могли связываться со своими рецепторами и вызывать такую же — или гораздо более сильную — реакцию.
Проще говоря, агонист подобен ключу, который вставляется в замок (рецептор) и поворачивает его, чтобы открыть дверь (или послать биохимический или электрический сигнал для воздействия).Естественный агонист — это главный ключ, но можно разработать другие ключи (агонистические препараты), которые будут выполнять ту же работу.
Например,Морфин не был создан организмом, но его естественным образом можно найти в опийном маке. По счастливой случайности он имитирует форму естественных агонистов опиоидов, эндорфинов, которые являются естественными болеутоляющими, ответственными за «высокий уровень эндорфина».
Специфические эффекты, такие как облегчение боли или эйфория, происходят потому, что опиоидные рецепторы присутствуют только в некоторых частях мозга и тела, которые влияют на эти функции.
Основной активный ингредиент каннабиса, ТГК, является агонистом каннабиноидного рецептора, а галлюциногенный препарат ЛСД представляет собой синтетическую молекулу, имитирующую агонистическое действие нейромедиатора серотонина на одном из его многочисленных рецепторов — рецепторе 5HT2A.
CC BY-NDПрепараты-антагонисты
Антагонист — это лекарство, предназначенное для прямого противодействия действию агониста.
Опять же, используя аналогию с замком и ключом, антагонист подобен ключу, который хорошо входит в замок, но не имеет правильной формы, чтобы повернуть замок.Когда этот ключ (антагонист) вставлен в замок, правильный ключ (агонист) не может войти в тот же замок.
Таким образом, действие агониста блокируется присутствием антагониста в молекуле рецептора.
И снова давайте представим морфин как агонист опиоидных рецепторов. Если кто-то переживает потенциально смертельную передозировку морфина, антагонист опиоидных рецепторов налоксон может обратить вспять эффекты.
Это связано с тем, что налоксон (продаваемый как Narcan) быстро захватывает все опиоидные рецепторы в организме и предотвращает связывание морфина и их активацию.
Морфин отскакивает от рецептора за секунды. Когда он не связан с рецептором, антагонист может проникнуть внутрь и заблокировать его. Поскольку рецептор не может быть активирован, когда антагонист занимает рецептор, реакции нет.
Эффекты Наркана могут быть драматичными. Даже если жертва передозировки находится без сознания или при смерти, она может полностью прийти в сознание и бодрствовать в течение нескольких секунд после инъекции.
CC BY-NDИнгибиторы мембранного транспорта
Мембранные переносчики — это большие белки, встроенные в клеточную мембрану, которые переносят более мелкие молекулы, такие как нейротрансмиттеры, из-за пределов клетки, которая их высвобождает, обратно внутрь.Некоторые препараты подавляют их действие.
Селективные ингибиторы обратного захвата серотонина (СИОЗС), такие как антидепрессант флуоксетин (прозак), работают следующим образом.
Серотонин — нейромедиатор мозга, регулирующий настроение, сон и другие функции, такие как температура тела. Он высвобождается из нервных окончаний, связываясь с рецепторами серотонина в соседних клетках мозга.
Для того, чтобы процесс работал гладко, мозг должен быстро выключить сигналы, исходящие от серотонина, вскоре после того, как химические вещества высвободятся из терминалов.В противном случае мгновенный контроль над мозгом и функциями тела был бы невозможен.
Мозг делает это с помощью переносчиков серотонина в мембране нервных окончаний. Подобно пылесосу, транспортеры собирают молекулы серотонина, которые не связаны с рецепторами, и транспортируют их обратно внутрь терминала для дальнейшего использования.
ПрепаратыSSRI застревают внутри вакуумного шланга, поэтому несвязанные молекулы серотонина не могут быть доставлены обратно в терминал.
Поскольку тогда большее количество молекул серотонина остается вокруг рецепторов дольше, они продолжают их стимулировать.
Можно грубо сказать, что дополнительный серотонин умеренно увеличивает громкость сигнала, чтобы улучшить позитивное настроение. Но реальный способ, которым это влияет на депрессию и тревогу, намного сложнее.
Около 40% всех лекарственных препаратов нацелены только на одно суперсемейство рецепторов — рецепторы, связанные с G-белком. Существуют вариации этих лекарственных механизмов, включая частичные агонисты и те, которые действуют как антагонисты, но несколько иначе.В целом, однако, многие лекарственные препараты попадают в описанные выше категории.
Что такое антагонист опиоидов?
В вашем организме есть опиоидная система, состоящая из рецепторов, которые взаимодействуют с естественными опиоидными соединениями (эндогенными опиоидами). Опиоиды активируют рецепторы, которые затем влияют на ваше настроение и способы обработки боли. Антагонисты опиоидов обращают действие опиоидов на противоположное, блокируя опиоидные рецепторы. Это руководство объяснит, как действуют антагонисты опиоидов и их использование в медицине.
Опиоидные агонисты, частичные агонисты и антагонисты: обзор
Опиоиды и родственные им препараты можно разделить на четыре категории в зависимости от их взаимодействия с опиоидными рецепторами:
- Полные агонисты: Опиоидный агонист связывается с опиоидными рецепторами и активирует их функции. Агонисты могут прикрепляться к опиоидным рецепторам в различной степени. Полные агонисты прочно прикрепляются к опиоидным рецепторам, производя одни из самых сильных эффектов среди всех опиоидных препаратов.Опиоиды, такие как фентанил, героин, морфин и оксикодон, считаются полными агонистами.
- Частичные агонисты: Частичные агонисты активируют опиоидные рецепторы в меньшей степени, чем полные агонисты. Хотя частичные агонисты могут иметь эффекты, аналогичные действию полных агонистов, они теряют эффективность при более высоких дозах. Опиоиды, которые относятся к категории частичных агонистов, включают бупренорфин и трамадол.
- Антагонисты: Антагонисты опиоидов связываются с опиоидными рецепторами, чтобы блокировать эффекты опиоидов.В отличие от опиоидных агонистов, они не вызывают эйфорического эффекта и не облегчают боль. Однако, поскольку они все еще связываются с рецепторами, они блокируют космические опиоиды, которые обычно уходят.
- Смешанные агонисты / антагонисты: Опиоидная система включает несколько типов рецепторов, которые по-разному реагируют на опиоиды. Смешанная активность агонистов / антагонистов зависит от типа рецептора. Он может действовать как агонист для одного типа рецептора, работая как антагонист для рецептора другого типа.
Поскольку опиоидная система управляет такими важными функциями, как боль и настроение, эти соединения влияют на работу вашего тела.
Как антагонисты опиоидов взаимодействуют с мозгом?
Антагонист опиоидов действует на человека, в организме которого присутствуют опиоиды. Люди, которые принимают антагонист без приема опиоидов, практически не испытывают никаких эффектов.
Однако, когда кто-то с опиоидами в организме принимает антагонист, антагонист сводит на нет большинство эффектов опиоидов.В некоторых случаях антагонист опиоидов может вызвать симптомы отмены, поскольку он блокирует доступ рецепторов к опиоидам.
О препаратах-антагонистах опиоидов
К двум наиболее распространенным препаратам-антагонистам опиоидов относятся:
- Налоксон: Вы можете найти налоксон в Suboxone®, лекарстве, используемом для лечения опиоидной зависимости в качестве меры, препятствующей злоупотреблению. Налоксон также используется в качестве лекарственного средства, спасающего от передозировки опиоидов, и широко известен под торговой маркой «Наркан».
- Налтрексон: Некоторые люди, получающие лечение от опиоидной зависимости, принимают налтрексон, чтобы блокировать действие опиоидов. В результате они не чувствуют воздействия опиоидов в случае рецидива.
Эти лекарства помогают многим людям с опиоидной зависимостью управлять своими симптомами или выздоравливать после передозировки. Медицинские работники и немедицинские специалисты могут использовать налоксон, чтобы спасти жизнь человека, перенесшего передозировку опиоидов.
Узнайте больше об агонистах и антагонистах опиоидов в ресурсных центрах здравоохранения
Для получения дополнительной информации по темам, связанным с опиоидной зависимостью, посетите нашу страницу блога.Вы также можете связаться с нашей командой онлайн, чтобы записаться на прием в одну из наших клиник в Новой Англии.
Создание агонистов и антагонистов пептидных рецепторов
Rudinger, J. in Drug Design Vol. 2 (изд. Ariens, J.) 319–419 (Academic, New York, 1971).
Google ученый
Hruby, V.J. в статье Topics in Molecular Pharmacology (eds Burgen, A.SV & Roberts, G.К. К.) 99–126 (Elsevier North Holland, Амстердам, 1981).
Google ученый
Кесслер, Х. Конформация и биологическая активность циклических пептидов. Angew. Chemie Int. Edn Engl. 21 , 512–521 (1982). В этой статье изложена основная стратегия и обоснование циклизации пептидного остова в пептидном дизайне.
Артикул Google ученый
Грубый, В.J. Конформационные ограничения биологически активных пептидов через группы боковой цепи аминокислот. Науки о жизни 31 , 189–199 (1982). Обрисовывает в общих чертах основную стратегию и обоснование циклизации от боковой цепи к боковой цепи в дизайне пептидов.
CAS Статья Google ученый
Hirschmann, R. et al. De novo разработка и синтез непептидных пептидомиметиков соматостатина с использованием β-D-глюкозы в качестве новой основы. J. Am. Chem. Soc. 115 , 12550–12568 (1993).
CAS Статья Google ученый
Ян Л. и др. Синтез и биологическая активность сильнодействующих пептидомиметиков, селективных в отношении подтипа 2 рецептора соматостатина. Proc. Natl Acad. Sci. США 95 , 10836–10841 (1998).
CAS Статья Google ученый
Пастернак, А.и другие. Сильные пероральные биодоступные агонисты соматостатина: хорошее всасывание достигается за счет циклизации основной цепи мочевины. Bioorg. Med. Chem. Lett. 9 , 491–496 (1999).
CAS Статья Google ученый
Liao, S. et al. De novo дизайн, синтез и биологическая активность высокоаффинных и селективных непептидных агонистов δ-опиоидных рецепторов. J. Med. Chem. 41 , 4767–4776 (1998).
CAS Статья Google ученый
Шендерович, М. Д., Ляо, С., Цянь, X. и Хруби, В. Дж. Трехмерная модель дельта-опиоидного фармакофора: сравнительное молекулярное моделирование пептидных и непептидных лигандов. Биополимеры 53 , 565–580 (2000).
CAS Статья Google ученый
Хруби, В. Дж., Аль-Обейди, Ф.И Казмиерски, В. М. Новые подходы к молекулярному дизайну рецептор-селективных пептидных лигандов: конформационные, топографические и динамические соображения. Biochemical J. 268 , 249–262 (1990). Описывает базовую стратегию интеграции конформационных и топографических соображений в дизайн биологически активного пептида-лиганда. Предоставляет некоторые ключевые ссылки для этого подхода.
CAS Статья Google ученый
Hirschmann, R.Лечебная химия в золотой век биологии. Уроки пептидных и стероидных исследований. Angew. Chemie Int. Edn Engl. 30 1278–1301 (1991).
Артикул Google ученый
Ризо, Дж. И Гираш, Л. М. Ограниченные пептиды: модели биоактивных пептидов и белковых субструктур. Annu. Rev. Biochem. 61 , 387–418 (1992). Предоставляет подробный обзор использования конформационных ограничений в биологически активных пептидах и белках со многими ключевыми ссылками.
CAS Статья Google ученый
Хруби, В. Дж. Конформационные и топографические соображения при разработке биологически активных пептидов. Биополимеры 33 , 1073–1082 (1993).
CAS Статья Google ученый
Маршалл, Г. Р. Иерархический подход к пептидомиметическому дизайну. Тетраэдр 49 , 3547–3555 (1993).
CAS Статья Google ученый
Фарли Д. П., Аббенанте Г. и Марч Д. Р. Макроциклические пептидомиметики — заставляют пептиды принимать биоактивные конформации. Curr. Med. Chem. 2 , 654–686 (1995).
Google ученый
Вермей П. и Блок Д. Новые пептидные и белковые препараты. Pharm. Мировая наука. 18 , 87–93 (1996).
Артикул Google ученый
Хруби, В. Дж. И Бальс, П. М. Конформационные и топографические соображения при разработке пептидомиметиков-агонистов на основе пептидных отведений. Curr. Med. Chem. 7 , 945–970 (2000).
CAS Статья Google ученый
Хруби, В. Дж. Дизайн в топографическом пространстве пептидов и пептидомиметических лигандов, которые влияют на поведение.Взгляд химика на проблему разума и тела. В соотв. Chem. Res. 34 389–397 (2001).
CAS Статья Google ученый
Шпигель А. М. Генетические основы эндокринного заболевания: мутации G-белков и рецепторов, связанных с G-белками, при эндокринных заболеваниях. J. Clin. Эндокринол. Метаб. 81 , 2434–2442 (1996).
CAS PubMed Google ученый
Саламон, З.и другие. Плазмонно-резонансные исследования связывания агонистов / антагонистов с человеческим δ-опиоидным рецептором: новое структурное понимание взаимодействий рецептор-лиганд. Biophys. J. 79 , 2463–2474 (2000).
CAS Статья Google ученый
Salamon, Z., Alves, I., Cowell, S., Hruby, VJ & Tollin, G. в Peptides: The Wave of the Future (eds Lebl, M. & Houghten, RA) 297 –298 (Am. Peptide Soc., Сан-Диего, 2001). Ссылки 20 и 21 описывают однозначную демонстрацию — с использованием нового биофизического метода спектроскопии плазменного волноводного резонанса (PWR или CPWR) — того, что агонисты, антагонисты и обратные агонисты лиганды взаимодействуют с GPCR, δ-опиоидным рецептором, с образованием различных стабильных конформационных рецепторов. состояния.
Google ученый
Рамачандран, Г. Н., Рамакришнан, К., Сасисекхаран, В. Стереохимия конфигураций полипептидных цепей. J. Mol. Биол. 7 , 95–99 (1963).
CAS Статья Google ученый
Рамачандран Г. Н. и Сасисекхаран В. Конформация полипептидов и белков. Adv. Protein Chem. 23 , 283–438 (1968). Предоставляет подробное обсуждение низкоэнергетических конформаций пептидов и белков. Жизненно важное значение в дизайне пептидов и белков.
CAS Статья Google ученый
Маршалл, Г.R. & Bosshard, H.E. Ангиотензин II. Биологически активный экстерьер. Circ. Res. 9 (Приложение 2), 143–150 (1972).
Google ученый
Toniolo, C. Конформационно ограниченные пептиды посредством короткодействующей циклизации. Внутр. J. Peptide Protein Res. 35 , 287–300 (1990).
CAS Статья Google ученый
Спатола, А.в Химия и биохимия аминокислот, пептидов и белков Vol. 7 (ред. Вайнштейн, Б.) 267–357 (Марсель Деккер, Нью-Йорк, 1983).
Google ученый
Хруби, В. Дж. И Гериг, С. А. Последние разработки в области создания рецептор-специфических опиоидных пептидов. Med. Res. Ред. 9 , 343–401 (1989).
CAS Статья Google ученый
Мосберг, Х.I. et al. Бис-пеницилламиновые энкефалины обладают значительно улучшенной специфичностью в отношении δ-опиоидных рецепторов. Proc. Natl Acad. Sci. США 80 , 5871–5874 (1983).
CAS Статья Google ученый
Weber, S.J. et al. Распространение и обезболивание [D-pen 2 , D-pen 5 ] энкефалина и двух галогенированных аналогов после внутривенного введения. J. Pharmacol. Exp. Терапия. 259 , 1109–1117 (1991).
CAS Google ученый
Williams, SA, Abbruscato, TJ, Hruby, VJ & Davis, TP Прохождение энкефалина, селективного к δ-опиоидным рецепторам, [D-пеницилламин 2,5 ] энкефалина, через кровь-мозг и кровь-спинномозговой жидкостные барьеры. J. Neurochem. 66 1289–1299 (1996).
CAS Статья Google ученый
Грубый, В.J., Kao, L. -F., Pettitt, BM & Karplus, M. Конформационные свойства δ-опиоидного пептида [D-pen 2 , D-pen 5 ] -энкефалина в водном растворе определены с помощью Расчеты ЯМР и минимизации энергии. J. Am. Chem. Soc. 110 , 3351–3359 (1988).
CAS Статья Google ученый
Флиппен-Андерсон, Дж. Л., Хруби, В. Дж., Коллинз, Н., Джордж, К. и Кадни, Б.Рентгеновская структура энкефалина [D-Pen 2 , D-pen 5 ], высокоактивного соединения, селективного к δ-опиоидным рецепторам: сравнение с предлагаемыми конформациями раствора. J. Am. Chem. Soc. 116 , 7523–7531 (1994).
CAS Статья Google ученый
Шендерович, М. Д., Ляо, С., Цянь, X. и Хруби, В. Дж. Трехмерная модель дельта-опиоидного фармакофора: сравнительное молекулярное моделирование пептидных и непептидных лигандов. Биополимеры 53 , 565–580 (2000).
CAS Статья Google ученый
Hruby, V.J., Li, G., Haskell-Luevano, C. & Shenderovich, M.D. Дизайн пептидов, белков и пептидомиметиков в пространстве Хи. Биополимеры 43 , 219–266 (1997). Обзор и обсуждение аминокислотной топографии, синтеза топографически ограниченных аминокислот и топографического дизайна.Дает подробное обоснование ограничений боковой цепи аминокислот при углах χ и их значение для биологической активности пептидов и белков. Имеет решающее значение для дизайна биологически активных пептидов и протеиновых «активных центров».
CAS Статья Google ученый
Гибсон С. Э., Гильо Н. и Тозер М. Дж. К управлению χ-пространством: конформационно ограниченные аналоги Phe, Tyr, Trp и His. Тетраэдр 55 , 585–615 (1999).
CAS Статья Google ученый
Qian, X. et al. Исследование стереохимических требований к распознаванию рецепторов δ-опиоидных агонистов посредством топографических модификаций в позиции 1. J. Am. Chem. Soc. 118 , 7280–7290 (1996)
CAS Статья Google ученый
Бильский, Е.J., Qian, X., Hruby, VJ & Porreca, F. Антиноцицептивная активность [β-метил-2 ‘, 6’-диметилтирозин 1 ] -замещенного циклического- [D-Pen 2 , D-pen 5 ] энкефалина и аналогов [D-Ala 2 , Asp 4 ] дельторфина. J. Pharmacol. Exp. Терапия. 293 , 151–158 (2000).
CAS Google ученый
Хруби В. Дж. Стратегии разработки пептидных антагонистов. Прог. Brain Res. 92 , 215–224 (1992).
CAS Статья Google ученый
Коуэлл, С. М., Бальсе-Сринивасан, П. М., и Ан, Дж. -М. И Hruby, V.J. Дизайн и синтез пептидных антагонистов и обратных агонистов для рецепторов, связанных с G-белком. Methods Enzymol. 343 , 49–72 (2002).
Артикул Google ученый
Терретт, Н.K. Combinatorial Chemistry (Oxford Univ. Press, Oxford, 1998).
Google ученый
Hruby, V.J. in Практика медицинской химии Ch. 9 (изд. Вермут, К. Г.) 135–151 (Academic, Лондон, 1996).
Google ученый
Yu, J. & Smith, G.P. Созревание аффинности фаговых пептидных лигандов. Methods Enzymol. 267 , 3–27 (1996).
CAS Статья Google ученый
Schwyzer, R. ACTH: краткий вводный обзор. Ann. Акад. Sci. 297 , 3–25 (1977).
CAS Статья Google ученый
Ямаширо, Д. и Ли, К. Х. в статье Пептиды: анализ, синтез, биология Vol. 6 .(ред. Уденфренд, С. и Мейенхофер, Дж.) 191–217 (Academic, New York, 1984).
Google ученый
Ли, Н. М. и Смит, А. П. Белок-липидная модель опиатного рецептора. Life Sci. 26 , 1459–1464 (1980).
CAS Статья Google ученый
Гарзия, Р., Ямаширо, Д., Хаммондс, Г. Р. Младший, Джи, Дж. И Ли, К. Х. Синтез и свойства человеческого β-эндорфина (1–17) и его аналогов. Внутр. J. Peptide Protein Res. 19 , 432–435 (1982).
CAS Статья Google ученый
Ferrara, P. & Li, C. H. Взаимодействие β-эндорфина синтетических аналогов, имеющих разную длину цепи, с рецепторами морфина и энкефалина в мембранах мозга крыс. Внутр. J. Peptide Protein Res. 19 , 259–262 (1982).
CAS Статья Google ученый
Грубый, В.Дж., Райт, Д. Э., Лин, М. С. и Родбелл, М. Полусинтетические производные глюкагона для структурно-функциональных исследований. Метаболизм 25 (Приложение 1), 1323–1325 (1976).
CAS Статья Google ученый
Брегман, М. Д., Триведи, Д. Б. и Хруби, В. Дж. Аминогруппы глюкагона: оценка модификаций, ведущих к антагонизму и агонизму. J. Biol. Chem. 255 , 11725–11733 (1980).
CAS PubMed Google ученый
Джонсон, Д. Г., Гебель, К. У., Хруби, В. Дж., Брегман, М. Д. и Триведи, Д. Б. Снижение гипергликемии у диабетических крыс с помощью антагониста рецепторов глюкагона. Наука 215 , 1115–1116 (1982).
CAS Статья Google ученый
Azizeh, B. Y. et al. [DesHis 1 , desPhe 6 , Glu 9 ] амид глюкагона: новый «чистый» антагонист глюкагона. Bioorg. Med. Chem. Lett. 5 , 1849–1852 (1995).
CAS Статья Google ученый
Hruby, V.J., Ahn, J. -M. И Триведи, Д. Дизайн и биологическая активность агонистов и антагонистов глюкагона, а также их использование при изучении механизмов действия глюкозы. Curr. Med. Chem. 1 , 199–215 (2001).
CAS Статья Google ученый
Унсон, К.Г., Андреу, Д., Гургенда, Э. М. и Меррифилд, Р. Б. Синтетические пептидные антагонисты глюкагона. Proc. Natl Acad. Sci. США 84 4083–4087 (1987).
CAS Статья Google ученый
Van Tine, B.A. et al. Анализ накопления циклического аденозина 3 ‘, 5’-монофосфата на низком уровне [DesHis 1 , desPhe 6 , Glu 9 ] глюкагон-Nh3 идентифицирует антагонисты глюкагона из слабых частичных агонистов / антагонистов. Эндокринология 137 , 3316–3322 (1996).
CAS Статья Google ученый
Hruby, V.J., Chow, M. -S. И Смит, Д. Д. Конформационные и структурные аспекты связывания окситоцина с рецептором и биологической активности. Annu. Rev. Pharmacol. Toxicol. 30 , 501–534 (1990).
CAS Статья Google ученый
Лебль, М.in Handbook of Neurohypophyseal Hormone Analogs Vol. 2 Часть 1 (ред. Йост, К., Лебл, М. и Бртник, Ф.) 17–74 (CRC, Boca Raton, 1987).
Google ученый
Manning, M. et al. in Peptides 1988 (под ред. Jung, G. & Bayer, E.) 552–555 (Walter de Guyter, Berlin, 1989).
Hill, P. S., Smith, D. D., Slaninová, J. & Hruby, V. J. Бициклизация слабого агониста окситоцина дает очень мощный антагонист окситоцина. J. Am. Chem. Soc. 112 3110–3113 (1990).
CAS Статья Google ученый
Meraldi, J. -P., Hruby, V.J. & Brewster, A. I. R. Относительная конформационная жесткость окситоцина и [1-пеницилламин] -окситоцина: предложение о связи конформационной гибкости с агонизмом и антагонизмом пептидного гормона. Proc. Natl Acad. Sci. США 74 , 1373–1377 (1977).
CAS Статья Google ученый
Вуд, С.P. et al. анализ кристаллической структуры деаминоокситоцина: конформационная гибкость и связывание с рецептором. Science 232 , 633–636 (1986).
CAS Статья Google ученый
Хруби, В. Дж. Влияние рентгеновской структуры деаминоокситоцина на взаимодействия агонист / антагонист-рецептор. Trends Pharmacol. Sci. 8 336–339 (1987).
CAS Статья Google ученый
Шендерович, М.Д., Кёвер, К. Э., Вилке, С., Коллинз, Н. и Хруби, В. Дж. Конформации раствора мощных бициклических антагонистов окситоцина с помощью спектроскопии ядерного магнитного резонанса и моделирования молекулярной динамики. J. Am. Chem. Soc. 119 5833–5846 (1997).
CAS Статья Google ученый
Liao, S. et al. Замена ограниченных боковой цепью аминокислот α-метил-2 ‘, 6′-диметил-4’-метокситирозина в положении 2 аналога бициклического окситоцина обеспечивает уникальное понимание биоактивной топографии антагонистов окситоцина. J. Am. Chem. Soc. 120 , 7393–7394 (1998).
CAS Статья Google ученый
Cho, N. et al. Открытие нового, мощного и перорально активного непептидного антагониста рецептора человеческого лютеинизирующего гормона (LHRH). J. Med. Chem. 41 , 4190–4195 (1998).
CAS Статья Google ученый
Чо, Н.и другие. Тиено [2,3-d] пиримидин-3-уксусные кислоты. Новый класс антагонистов непептидных рецепторов эндотелина. Chem. Pharm. Бык. 46 , 1724–1737 (1998).
CAS Статья Google ученый
Генри, Дж. А., Хоруэлл, Д. К., Мичем, К. Г. и Рис, Д. С. Исследование сродства структуры боковых цепей аминокислот в нейротензине: N- и C-концевые делеции и Ala-сканирование. Bioorg. Med. Chem. Lett. 3 , 949–952 (1993).
CAS Статья Google ученый
Sawyer, T. K. et al. [Nle 4 , D-Phe 7 ] -α-меланоцит-стимулирующий гормон: высокоэффективный α-меланотропин со сверхдлительной биологической активностью. Proc. Natl Acad. Sci. США 77 , 5754–5758 (1980).
CAS Статья Google ученый
Сойер Т.К., Хруби, В. Дж., Дарман, П. С. и Хэдли, М. Е. [4-полуцистин, 10-полуцистин] -α-меланоцит-стимулирующий гормон: циклический α-меланотропин, проявляющий биологическую активность суперагонистов. Proc. Natl Acad. Sci. США 79 , 1751–1755 (1982).
CAS Статья Google ученый
Ан, Дж. -М. и другие. Новый подход к поиску биоактивной конформации глюкагона: позиционное циклизационное сканирование. J. Med. Chem. 44 , 3109–3116 (2001).
CAS Статья Google ученый
Агонист — определение, типы и тест
Определение агониста
Агонист — это молекула, которая может связывать и активировать рецептор, чтобы вызвать биологическую реакцию. Активности, опосредованной агонистами, противостоят антагонисты, которые ингибируют биологический ответ, вызванный агонистом. Уровень агониста, необходимый для индукции желаемого биологического ответа, называется эффективностью.Эффективность агониста определяется путем измерения концентрации агониста, необходимой для индукции половины максимального ответа, называемого значением EC 50 . Следовательно, агонисты с большей эффективностью будут иметь меньшие значения EC 50 . Активность агонистов часто рассчитывается в фармацевтической промышленности, так как дозировка лекарств, которые действуют как агонисты, зависит от EC 50 . На приведенной ниже диаграмме показано различие между агонистами природного происхождения, эффективностью агонистов лекарственных средств и ингибированием эффектов агонистов с помощью антагонистов.
Типы агонистов
Существует несколько типов агонистов, которые включают эндогенные, экзогенные, физиологические, суперагонисты, полные, частичные, обратные, необратимые, селективные и коагонисты. Каждый тип агониста обладает разными характеристиками и опосредует различную биологическую активность.
Эндогенные и экзогенные агонисты
Эндогенные агонисты представляют собой внутренние факторы, которые вызывают биологический ответ. Некоторые примеры эндогенных агонистов включают гормоны и нейротрансмиттеры, которые связываются с определенными рецепторами и вызывают желаемый ответ.Напротив, экзогенные агонисты представляют собой внешние факторы, которые связываются с различными рецепторами и вызывают биологический ответ. Примером экзогенного агониста является лекарство, такое как синтетический дофамин, которое связывается с рецептором дофамина и вызывает реакцию, аналогичную эндогенной передаче сигналов дофамина.
Физиологические агонисты
Физиологические агонисты — это агонисты, которые могут вызывать такой же биологический ответ; однако они не связываются с одним и тем же рецептором. Примером этого типа агониста является активация NF-каппа B как цитокинами (интерлейкин [IL] -6, IL-1 и фактор некроза опухоли), так и стимулами окружающей среды (например.g., бактериальные липополисахариды) посредством передачи сигналов через ассоциированные рецепторы цитокинов и рецепторы распознавания патогенов, соответственно.
Суперагонисты
Суперагонист — это агонист, способный вызывать биологический ответ, превышающий эффект, возникающий при связывании эндогенного агониста с рецептором. Самая распространенная форма суперагонистов — наркотики. Например, TGN1412 является суперагонистом CD28, что приводит к поликлональной активации Т-клеток и связано с риском продукции патогенных цитокинов при использовании в высоких дозах.
Полные агонисты против частичных
Полные агонисты способны полностью связываться и активировать свой родственный рецептор, тем самым вызывая полный ответ, способный к этому рецептору. Напротив, частичные агонисты также связываются с родственным рецептором; однако они вызывают лишь частичную реакцию. Частичные агонисты полезны для лечения и предотвращения лекарственной зависимости, поскольку они вызывают аналогичный эффект, хотя и менее сильный и вызывающий привыкание. Примером может служить использование бупренорфина в качестве альтернативы опиатам (например,g., морфин), поскольку он лишь частично взаимодействует с опиоидными рецепторами, что снижает вероятность опиатной зависимости.
Обратные агонисты
Обратный агонист связывается с тем же рецептором, что и агонист; однако он вызывает биологический ответ, противоположный агонисту. Обратный агонист отличается от антагониста тем, что вместо простого ингибирования ответа агониста индуцируется противоположный ответ.
Необратимые агонисты
Необратимые агонисты — это агонисты, которые образуют постоянную связь с рецептором посредством образования ковалентных связей.Некоторые из наиболее хорошо изученных необратимых агонистов — это агонисты μ-опиоидных рецепторов, такие как налоксазон и оксиморфазон.
Селективные агонисты
Селективные агонисты специфичны для конкретного рецептора. Например, IFN-гамма является селективным агонистом рецептора IFN-гамма.
Коагонисты
Коагонист требует комбинации двух или более агонистов, чтобы вызвать конкретный биологический ответ. Например, активация инфицированных макрофагов с образованием оксида азота зависит от связывания бактериальных лигандов, IFN-гамма и TNF, с их соответствующими рецепторами.
Тест
1. Агонист с высоким EC 50 связан с:
A. Для оптимального эффекта требуется высокая дозировка
B. Низкая эффективность
C. Низкая для оптимального эффекта требуется дозировка
D. Высокая эффективность
E. A и B
F. A и D
Ответ на вопрос № 1
E правильный. Лекарство с более высоким EC 50 будет иметь более низкую эффективность и, следовательно, потребует более высокой дозировки.
2. Молекула A связывается с рецептором A, а молекула B связывается с рецептором B. Оба агониста вызывают ответ C. Это пример агониста какого типа?
A. Обратные агонисты
B.